

This is a preprint.
Rapid protein evolution by few-shot learning with a protein language model
- PMID: 39071429
- PMCID: PMC11275896
- DOI: 10.1101/2024.07.17.604015
Rapid protein evolution by few-shot learning with a protein language model
Abstract
Directed evolution of proteins is critical for applications in basic biological research, therapeutics, diagnostics, and sustainability. However, directed evolution methods are labor intensive, cannot efficiently optimize over multiple protein properties, and are often trapped by local maxima. In silico-directed evolution methods incorporating protein language models (PLMs) have the potential to accelerate this engineering process, but current approaches fail to generalize across diverse protein families. We introduce EVOLVEpro, a few-shot active learning framework to rapidly improve protein activity using a combination of PLMs and protein activity predictors, achieving improved activity with as few as four rounds of evolution. EVOLVEpro substantially enhances the efficiency and effectiveness of in silico protein evolution, surpassing current state-of-the-art methods and yielding proteins with up to 100-fold improvement of desired properties. We showcase EVOLVEpro for five proteins across three applications: T7 RNA polymerase for RNA production, a miniature CRISPR nuclease, a prime editor, and an integrase for genome editing, and a monoclonal antibody for epitope binding. These results demonstrate the advantages of few-shot active learning with small amounts of experimental data over zero-shot predictions. EVOLVEpro paves the way for broader applications of AI-guided protein engineering in biology and medicine.
Figures
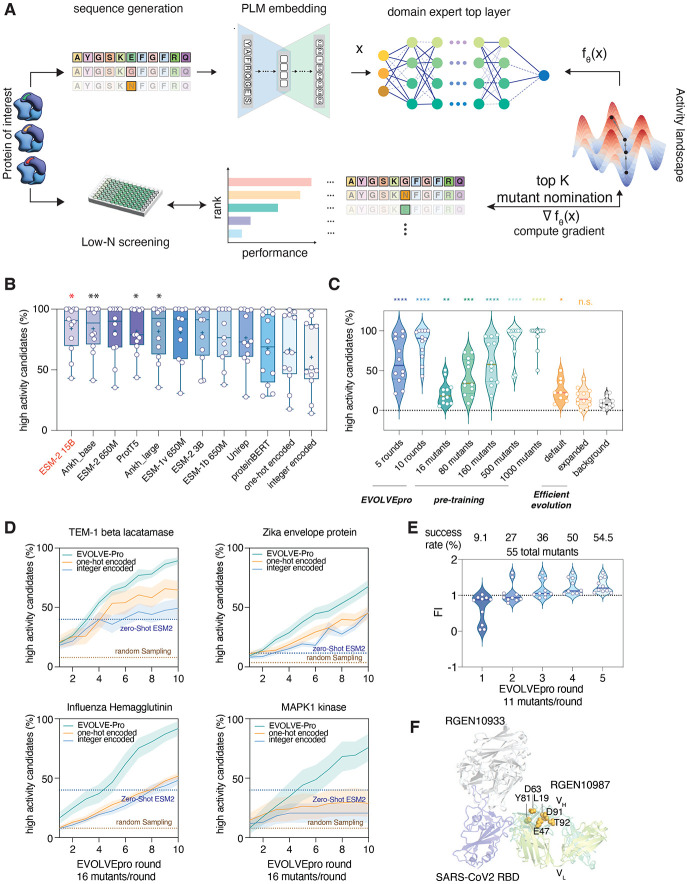
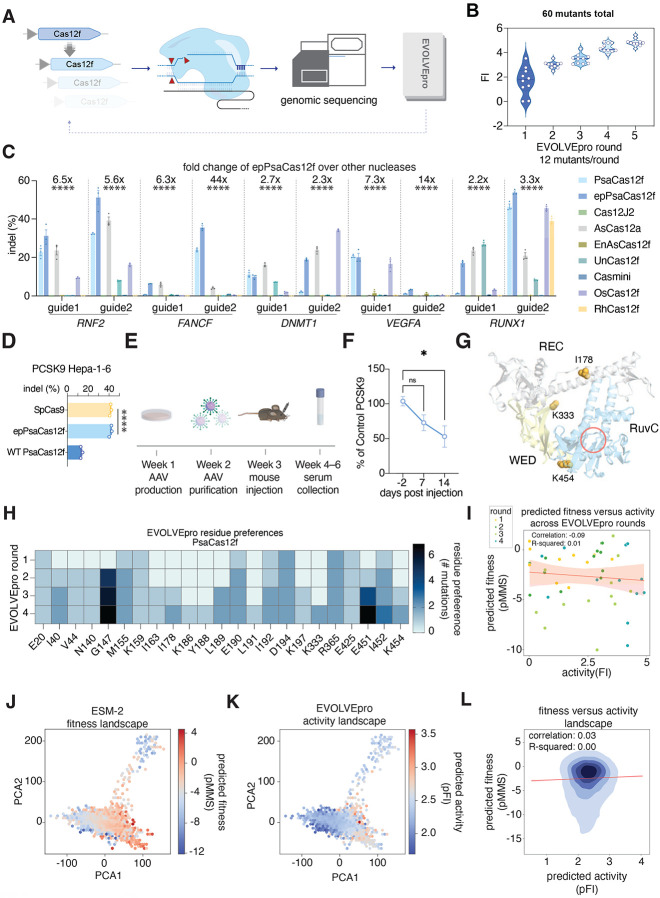




References
-
- Lin Z., Akin H., Rao R., Hie B., Zhu Z., Lu W., Smetanin N., Verkuil R., Kabeli O., Shmueli Y., Dos Santos Costa A., Fazel-Zarandi M., Sercu T., Candido S., Rives A., Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023). - PubMed
-
- Elnaggar A., Essam H., Salah-Eldin W., Moustafa W., Elkerdawy M., Rochereau C., Rost B., Ankh: Optimized Protein Language Model Unlocks General-Purpose Modelling, arXiv [cs.LG] (2023). http://arxiv.org/abs/2301.06568.
-
- He Y., Zhou X., Chang C., Chen G., Liu W., Li G., Fan X., Sun M., Miao C., Huang Q., Ma Y., Yuan F., Chang X., Protein language models-assisted optimization of a uracil-N-glycosylase variant enables programmable T-to-G and T-to-C base editing. Mol. Cell 84, 1257–1270.e6 (2024). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials