

Hepatocyte programmed cell death: the trigger for inflammation and fibrosis in metabolic dysfunction-associated steatohepatitis
- PMID: 39071804
- PMCID: PMC11272544
- DOI: 10.3389/fcell.2024.1431921
Hepatocyte programmed cell death: the trigger for inflammation and fibrosis in metabolic dysfunction-associated steatohepatitis
Abstract
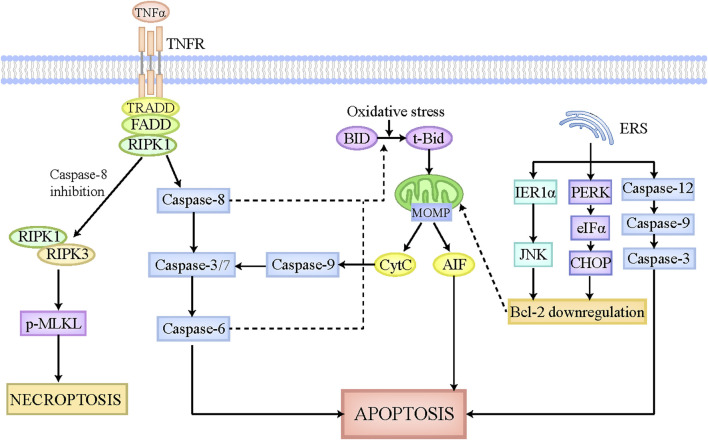
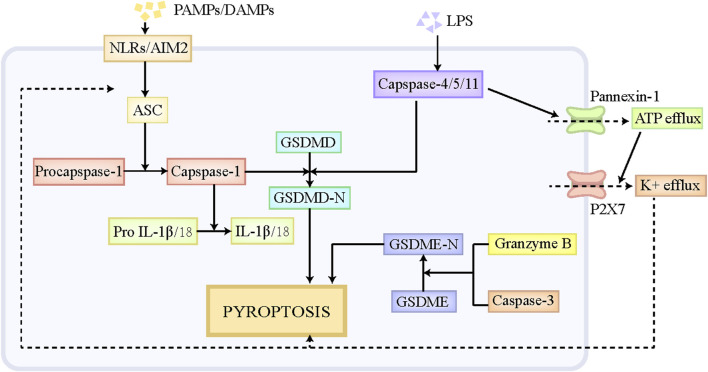
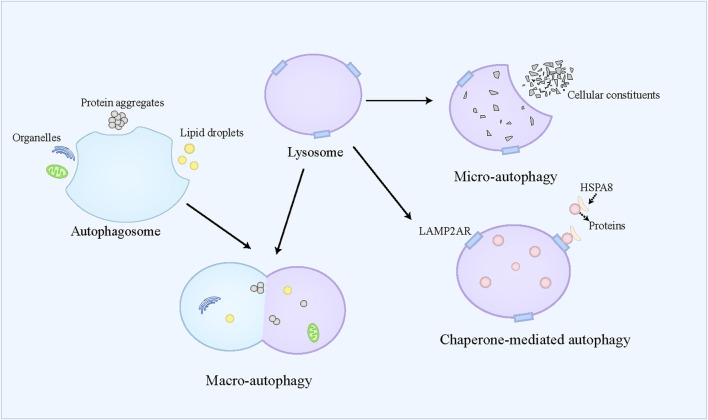
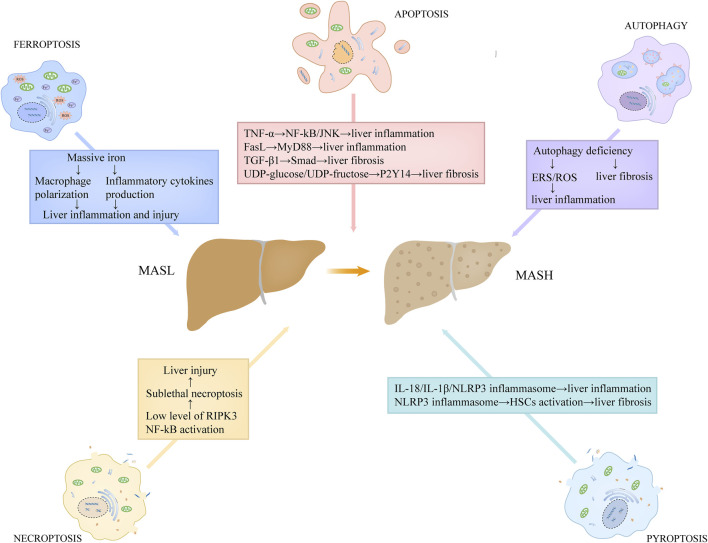
By replacing and removing defective or infected cells, programmed cell death (PCD) contributes to homeostasis maintenance and body development, which is ubiquitously present in mammals and can occur at any time. Besides apoptosis, more novel modalities of PCD have been described recently, such as necroptosis, pyroptosis, ferroptosis, and autophagy-dependent cell death. PCD not only regulates multiple physiological processes, but also participates in the pathogenesis of diverse disorders, including metabolic dysfunction-associated steatotic liver disease (MASLD). MASLD is mainly classified into metabolic dysfunction-associated steatotic liver (MASL) and metabolic dysfunction-associated steatohepatitis (MASH), and the latter putatively progresses to cirrhosis and hepatocellular carcinoma. Owing to increased incidence and obscure etiology of MASH, its management still remains a tremendous challenge. Recently, hepatocyte PCD has been attracted much attention as a potent driver of the pathological progression from MASL to MASH, and some pharmacological agents have been proved to exert their salutary effects on MASH partly via the regulation of the activity of hepatocyte PCD. The current review recapitulates the pathogenesis of different modalities of PCD, clarifies the mechanisms underlying how metabolic disorders in MASLD induce hepatocyte PCD and how hepatocyte PCD contributes to inflammatory and fibrotic progression of MASH, discusses several signaling pathways in hepatocytes governing the execution of PCD, and summarizes some potential pharmacological agents for MASH treatment which exert their therapeutic effects partly via the regulation of hepatocyte PCD. These findings indicate that hepatocyte PCD putatively represents a new therapeutic point of intervention for MASH.
Keywords: hepatic fibrosis; hepatic inflammation; metabolic disorders; metabolic dysfunction-associated steatohepatitis; programmed cell death.
Copyright © 2024 Cheng, Chu, Seki, Lin and Yang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Mechanism of PANoptosis in metabolic dysfunction-associated steatotic liver disease.Clin Res Hepatol Gastroenterol. 2024 Aug;48(7):102381. doi: 10.1016/j.clinre.2024.102381. Epub 2024 May 29. Clin Res Hepatol Gastroenterol. 2024. PMID: 38821484 Review.
-
Autophagy, Ferroptosis, Apoptosis and Pyroptosis in Metabolic Dysfunction-Associated Steatotic Liver Disease.Front Biosci (Landmark Ed). 2024 Jan 19;29(1):30. doi: 10.31083/j.fbl2901030. Front Biosci (Landmark Ed). 2024. PMID: 38287834 Review.
-
Sex-based differences in natural killer T cell-mediated protection against diet-induced steatohepatitis in Balb/c mice.Biol Sex Differ. 2023 Nov 14;14(1):85. doi: 10.1186/s13293-023-00569-w. Biol Sex Differ. 2023. PMID: 37964320 Free PMC article.
-
Targeting PYK2 with heterobifunctional T6BP helps mitigate MASLD and MASH-HCC progression.J Hepatol. 2025 Feb;82(2):277-300. doi: 10.1016/j.jhep.2024.08.029. Epub 2024 Sep 10. J Hepatol. 2025. PMID: 39260704
-
The Lasting Effects of COVID-19 on the Progression of Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD).Cureus. 2023 Sep 14;15(9):e45231. doi: 10.7759/cureus.45231. eCollection 2023 Sep. Cureus. 2023. PMID: 37842470 Free PMC article. Review.
Cited by
-
Metabolism of hepatic stellate cells in chronic liver diseases: emerging molecular and therapeutic interventions.Theranostics. 2025 Jan 2;15(5):1715-1740. doi: 10.7150/thno.106597. eCollection 2025. Theranostics. 2025. PMID: 39897543 Free PMC article. Review.
-
Targeting Metabolism: Innovative Therapies for MASLD Unveiled.Int J Mol Sci. 2025 Apr 25;26(9):4077. doi: 10.3390/ijms26094077. Int J Mol Sci. 2025. PMID: 40362316 Free PMC article. Review.
-
Unraveling the Metabolic Pathways Between Metabolic-Associated Fatty Liver Disease (MAFLD) and Sarcopenia.Int J Mol Sci. 2025 May 14;26(10):4673. doi: 10.3390/ijms26104673. Int J Mol Sci. 2025. PMID: 40429815 Free PMC article. Review.
-
Metabolic-Associated Fatty Liver Disease: The Influence of Oxidative Stress, Inflammation, Mitochondrial Dysfunctions, and the Role of Polyphenols.Pharmaceuticals (Basel). 2024 Oct 10;17(10):1354. doi: 10.3390/ph17101354. Pharmaceuticals (Basel). 2024. PMID: 39458995 Free PMC article. Review.
-
Melatonin Attenuates Ferritinophagy/Ferroptosis by Acting on Autophagy in the Liver of an Autistic Mouse Model BTBR T+Itpr3tf/J.Int J Mol Sci. 2024 Nov 23;25(23):12598. doi: 10.3390/ijms252312598. Int J Mol Sci. 2024. PMID: 39684310 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources