

Genome wide association study of clinical duration and age at onset of sporadic CJD
- PMID: 39079175
- PMCID: PMC11280162
- DOI: 10.1371/journal.pone.0304528
Genome wide association study of clinical duration and age at onset of sporadic CJD
Abstract
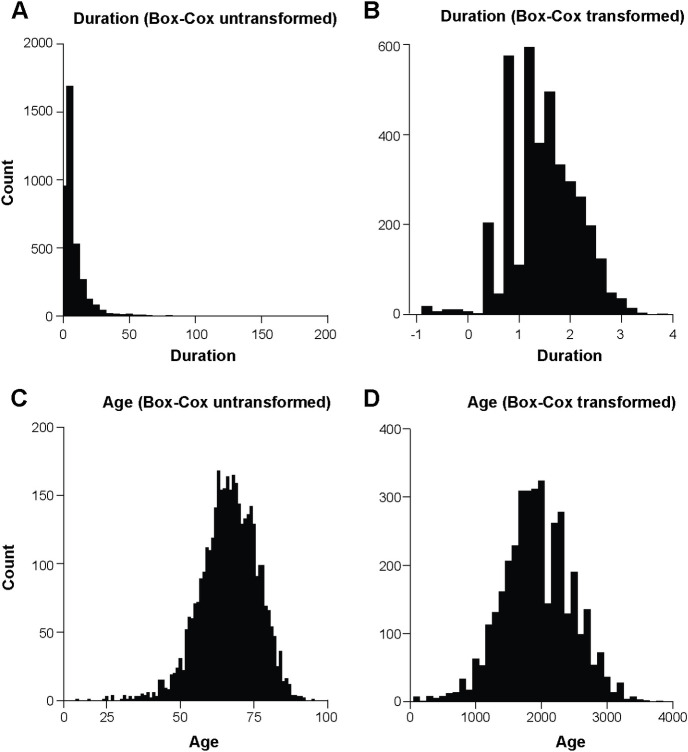
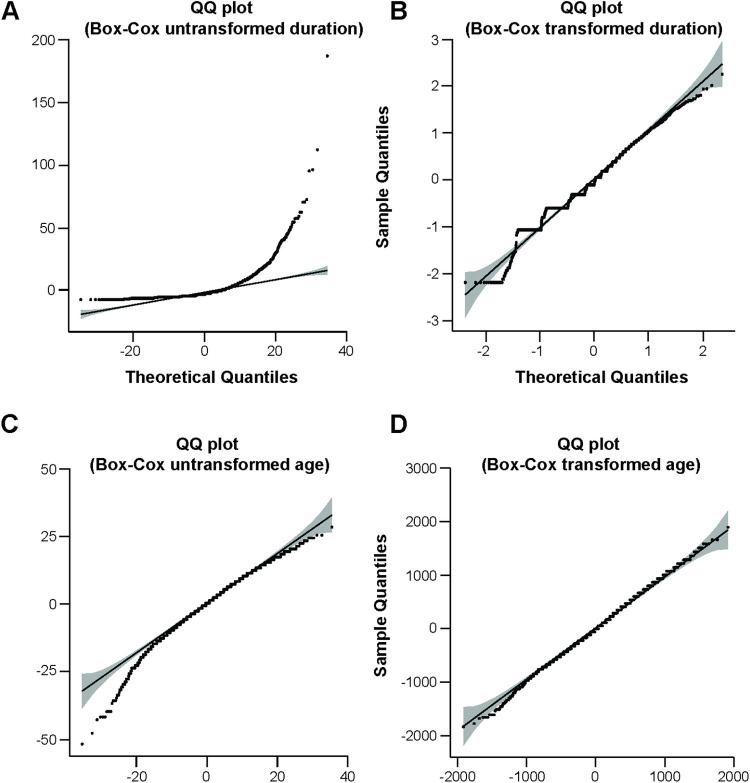
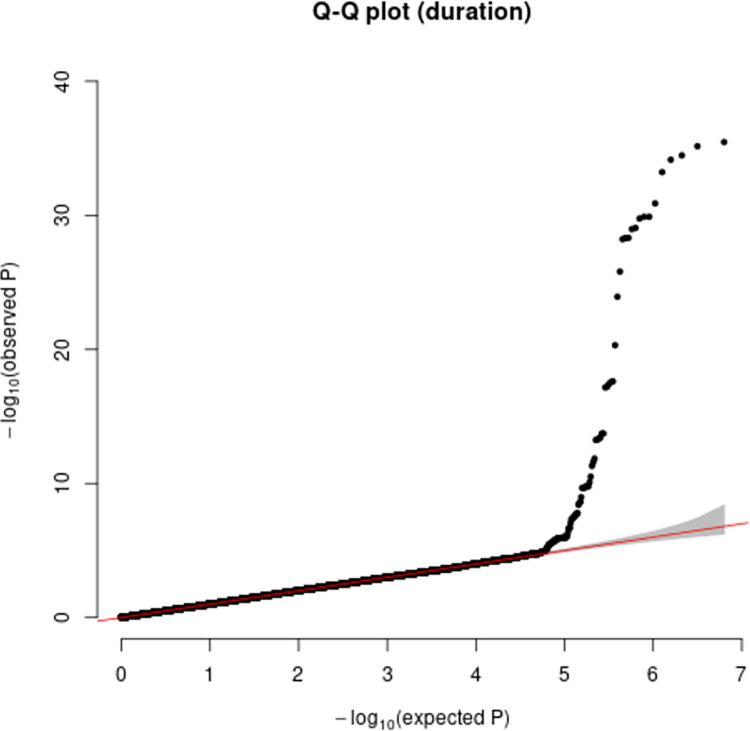
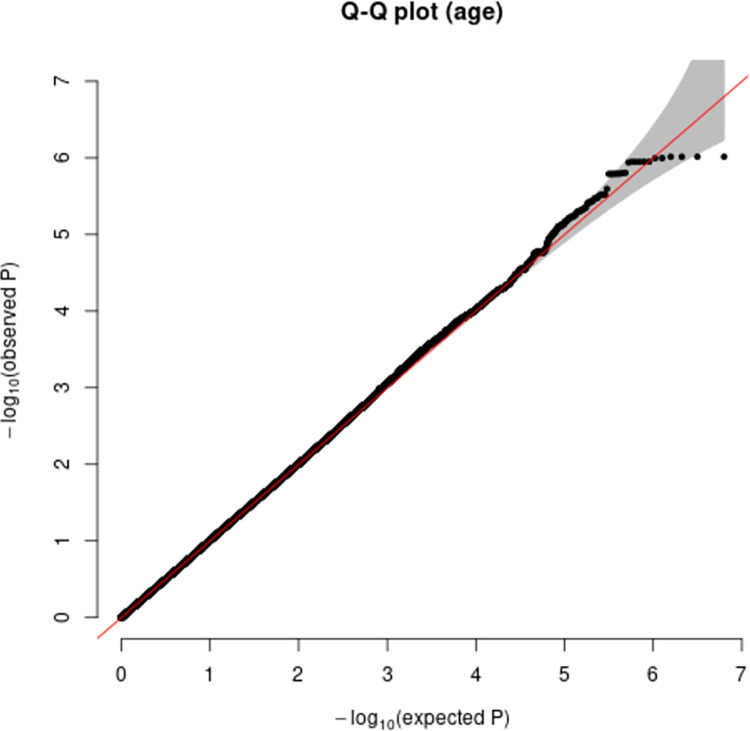
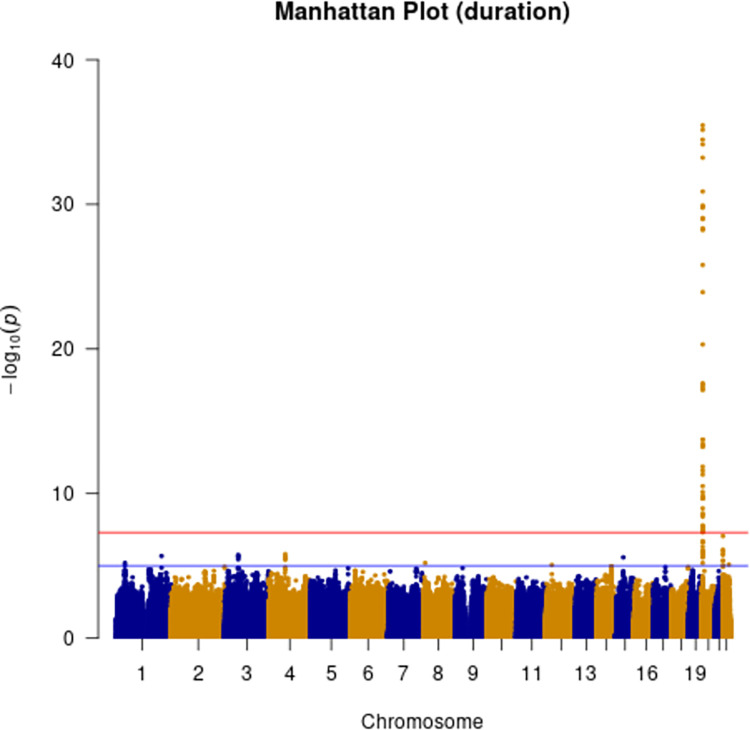
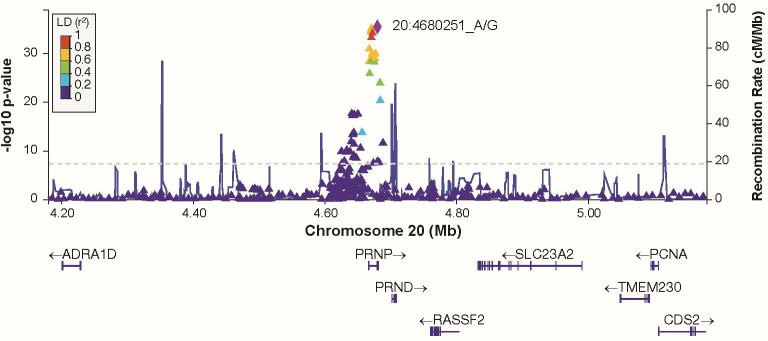
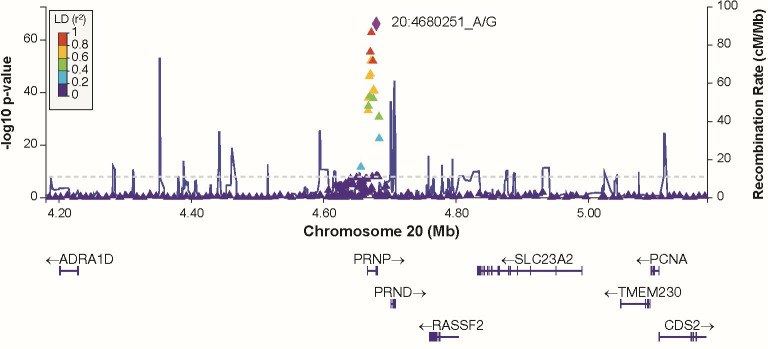
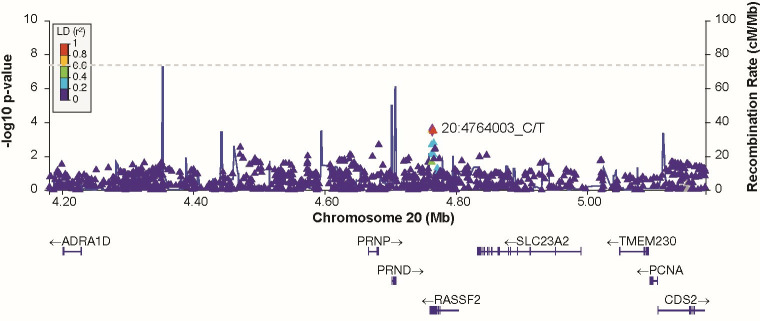
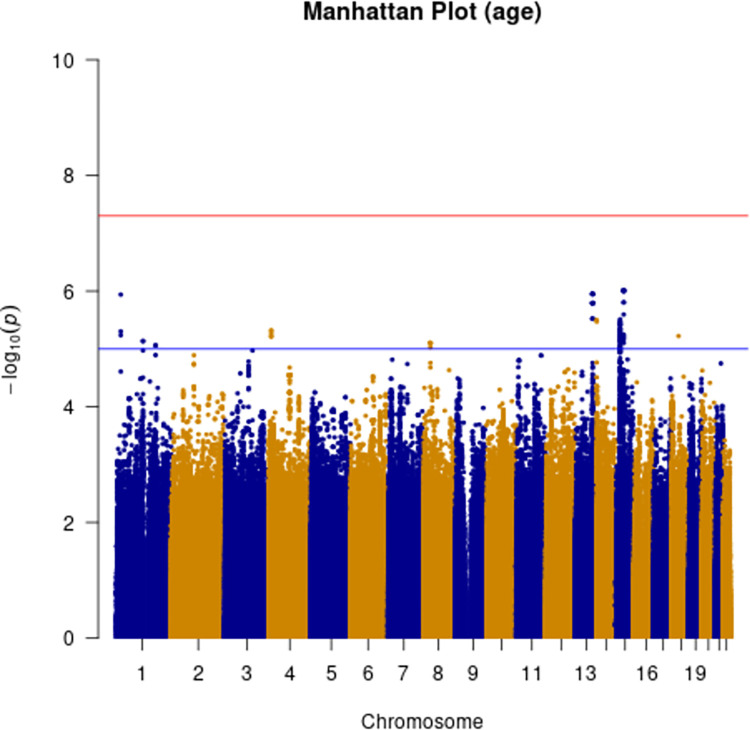
Human prion diseases are rare, transmissible and often rapidly progressive dementias. The most common type, sporadic Creutzfeldt-Jakob disease (sCJD), is highly variable in clinical duration and age at onset. Genetic determinants of late onset or slower progression might suggest new targets for research and therapeutics. We assembled and array genotyped sCJD cases diagnosed in life or at autopsy. Clinical duration (median:4, interquartile range (IQR):2.5-9 (months)) was available in 3,773 and age at onset (median:67, IQR:61-73 (years)) in 3,767 cases. Phenotypes were successfully transformed to approximate normal distributions allowing genome-wide analysis without statistical inflation. 53 SNPs achieved genome-wide significance for the clinical duration phenotype; all of which were located at chromosome 20 (top SNP rs1799990, pvalue = 3.45x10-36, beta = 0.34 for an additive model; rs1799990, pvalue = 9.92x10-67, beta = 0.84 for a heterozygous model). Fine mapping, conditional and expression analysis suggests that the well-known non-synonymous variant at codon 129 is the obvious outstanding genome-wide determinant of clinical duration. Pathway analysis and suggestive loci are described. No genome-wide significant SNP determinants of age at onset were found, but the HS6ST3 gene was significant (pvalue = 1.93 x 10-6) in a gene-based test. We found no evidence of genome-wide genetic correlation between case-control (disease risk factors) and case-only (determinants of phenotypes) studies. Relative to other common genetic variants, PRNP codon 129 is by far the outstanding modifier of CJD survival suggesting only modest or rare variant effects at other genetic loci.
Copyright: © 2024 Hummerich et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
Stéphane Haik reports grants from Santé Publique France, during the conduct of the study; grants from LFB Biomedicaments, grants from Institut de Recherche Servier, grants from MedDay Pharmaceuticals, outside the submitted work; In addition, Stéphane Haik has a patent Method for treating prion diseases (PCT/EP2019/070457) pending. Brian Appleby has received funding from CDC, NIH, CJD Foundation, Alector, and Ionis. He has served as a consultant for Ionis, Sangamo, and Gate Biosciences. He has received royalties from Wolter Kluwers. Karl Fronztek reports grants from Ono Pharmaceuticals outside the submitted work. Simon Mead reports grants from Medical Research Council (UK) and grants from National Institute of Health Research’s Biomedical Research Centre at University College London Hospitals NHS Foundation Trust during the conduct of the study. Gabor G Kovacs reports personal fees from Biogen, outside the submitted work. John Collinge reports grants from Medical Research Council, grants from NIHR UCLH Biomedical Research Centre, during the conduct of the study; and is a Director and shareholder of D-Gen Limited, an academic spinout in the field of prion disease diagnostics, decontamination and therapeutics. Inga Zerr reports grants from the Bundesministerium für Gesundheit via Robert Koch institute, JPND and personal fees (not related to the content of the manuscript) from Ferring Pharmaceuticals and IONIS, speaking honoraria for medical lectures from Lilly, Biogen, Medfora, DGLN (German Society for cerebrospinal fluid diagnostics in Neurology). Maurizio Pocchiari reports personal fees from Ferring Pharmaceuticals, personal fees from CNCCS (Collection of National Chemical Compounds and Screening Center), non-financial support from Fondazione Cellule Staminali, outside the submitted work. Michael D Geschwind has consulted for3D Communications, Adept Field Consulting, Advanced Medical Inc., Best Doctors Inc., Second Opinion Inc., Gerson Lehrman Group Inc., Guidepoint Global LLC, InThought Consulting Inc., Market Plus, Trinity Partners LLC, Biohaven Pharmaceuticals, Quest Diagnostics and various medical-legal consulting. He has received speaking honoraria for various medical center lectures and from Oakstone publishing. He has received past research support from Alliance Biosecure, CurePSP, the Tau Consortium, and Quest Diagnostics. Michael D Geschwind serves on the board of directors for San Francisco Bay Area Physicians for Social Responsibility and on the editorial board of Dementia & Neuropsychologia.
Figures
References
-
- Thompson AG, Lowe J, Fox Z, Lukic A, Porter MC, Ford L, et al. The Medical Research Council prion disease rating scale: a new outcome measure for prion disease therapeutic trials developed and validated using systematic observational studies. Brain. 2013; 136(Pt 4): 1116–1127. doi: 10.1093/brain/awt048 - DOI - PubMed
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
