

Cancer stem cells release interleukin-33 within large oncosomes to promote immunosuppressive differentiation of macrophage precursors
- PMID: 39079535
- PMCID: PMC11324407
- DOI: 10.1016/j.immuni.2024.07.004
Cancer stem cells release interleukin-33 within large oncosomes to promote immunosuppressive differentiation of macrophage precursors
Abstract
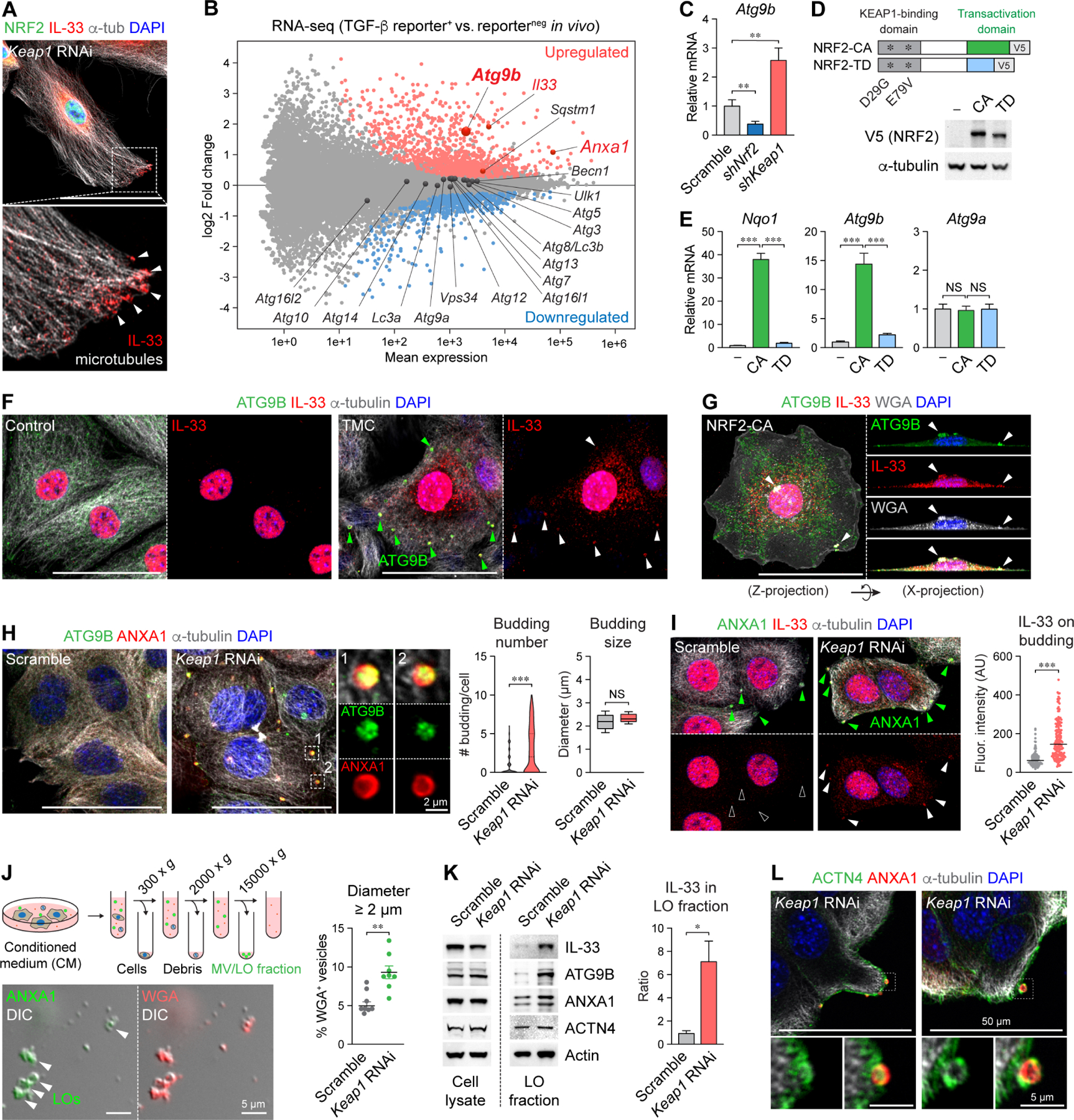
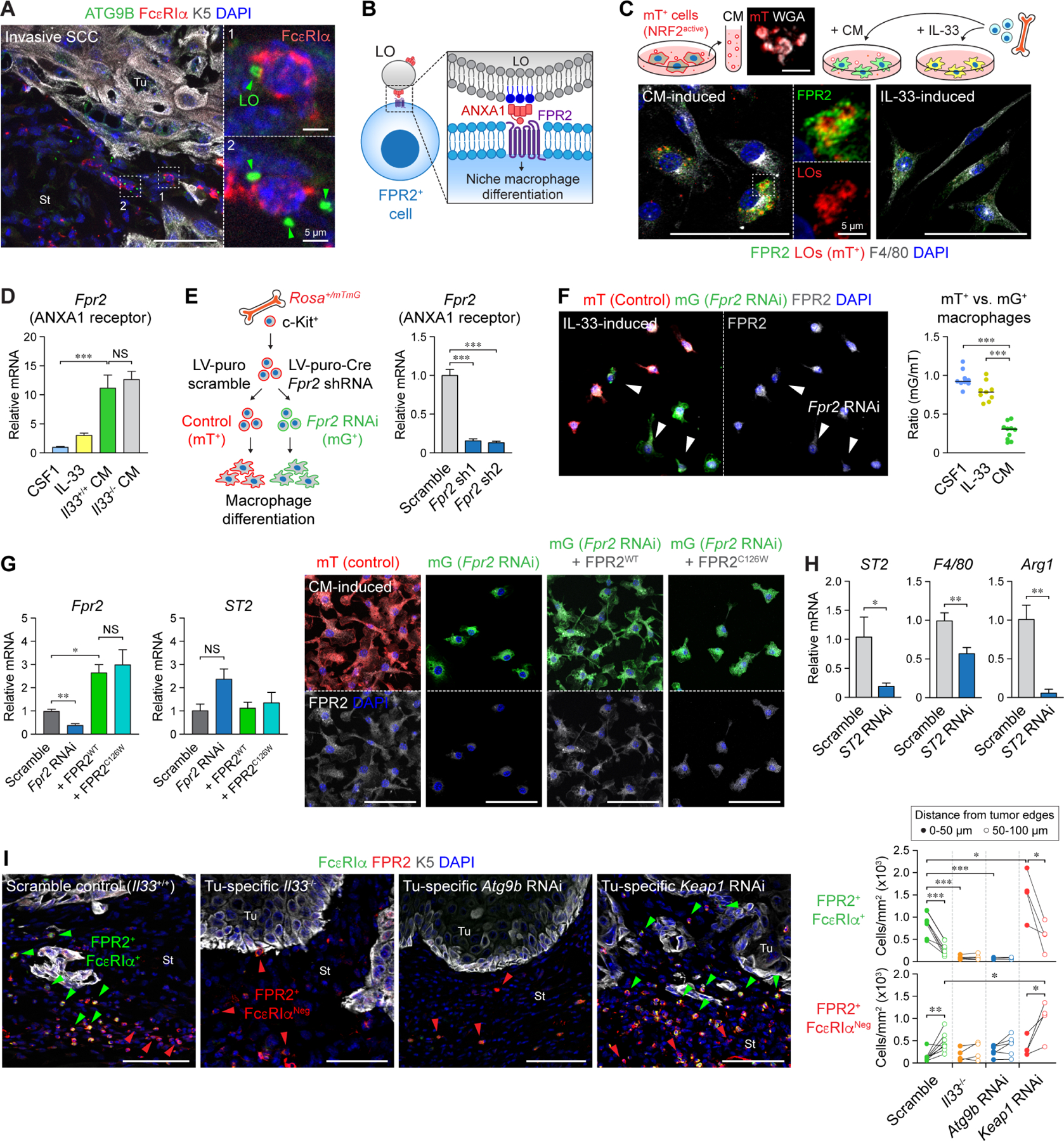
In squamous cell carcinoma (SCC), macrophages responding to interleukin (IL)-33 create a TGF-β-rich stromal niche that maintains cancer stem cells (CSCs), which evade chemotherapy-induced apoptosis in part via activation of the NRF2 antioxidant program. Here, we examined how IL-33 derived from CSCs facilitates the development of an immunosuppressive microenvironment. CSCs with high NRF2 activity redistributed nuclear IL-33 to the cytoplasm and released IL-33 as cargo of large oncosomes (LOs). Mechanistically, NRF2 increased the expression of the lipid scramblase ATG9B, which exposed an "eat me" signal on the LO surface, leading to annexin A1 (ANXA1) loading. These LOs promoted the differentiation of AXNA1 receptor+ myeloid precursors into immunosuppressive macrophages. Blocking ATG9B's scramblase activity or depleting ANXA1 decreased niche macrophages and hindered tumor progression. Thus, IL-33 is released from live CSCs via LOs to promote the differentiation of alternatively activated macrophage, with potential relevance to other settings of inflammation and tissue repair.
Keywords: ATG9b; FPR2; IL-33; annexin A1; cancer stem cell niche; cancer stem cells; large oncosomes; macrophages; squamous cell carcinoma; tumor microenvironment.
Copyright © 2024 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
