

pyRBDome: a comprehensive computational platform for enhancing RNA-binding proteome data
- PMID: 39079742
- PMCID: PMC11289467
- DOI: 10.26508/lsa.202402787
pyRBDome: a comprehensive computational platform for enhancing RNA-binding proteome data
Abstract
High-throughput proteomics approaches have revolutionised the identification of RNA-binding proteins (RBPome) and RNA-binding sequences (RBDome) across organisms. Yet, the extent of noise, including false positives, associated with these methodologies, is difficult to quantify as experimental approaches for validating the results are generally low throughput. To address this, we introduce pyRBDome, a pipeline for enhancing RNA-binding proteome data in silico. It aligns the experimental results with RNA-binding site (RBS) predictions from distinct machine-learning tools and integrates high-resolution structural data when available. Its statistical evaluation of RBDome data enables quick identification of likely genuine RNA-binders in experimental datasets. Furthermore, by leveraging the pyRBDome results, we have enhanced the sensitivity and specificity of RBS detection through training new ensemble machine-learning models. pyRBDome analysis of a human RBDome dataset, compared with known structural data, revealed that although UV-cross-linked amino acids were more likely to contain predicted RBSs, they infrequently bind RNA in high-resolution structures. This discrepancy underscores the limitations of structural data as benchmarks, positioning pyRBDome as a valuable alternative for increasing confidence in RBDome datasets.
© 2024 Chu et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures






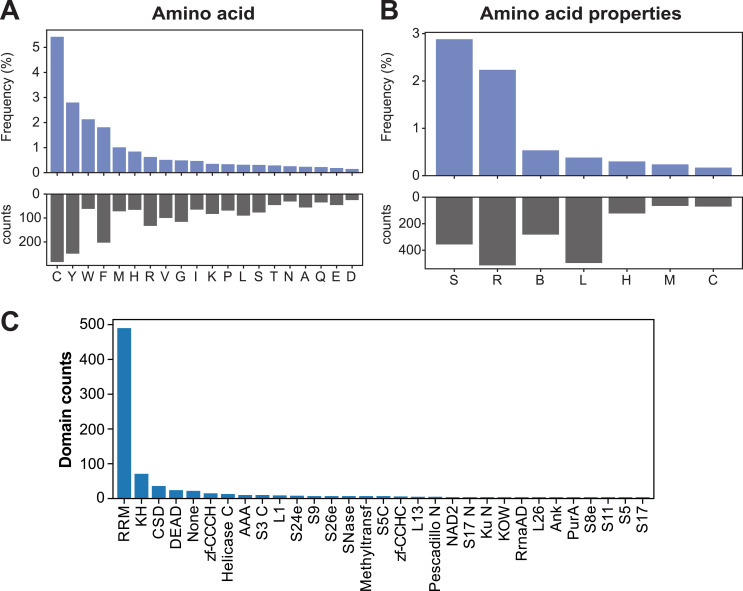










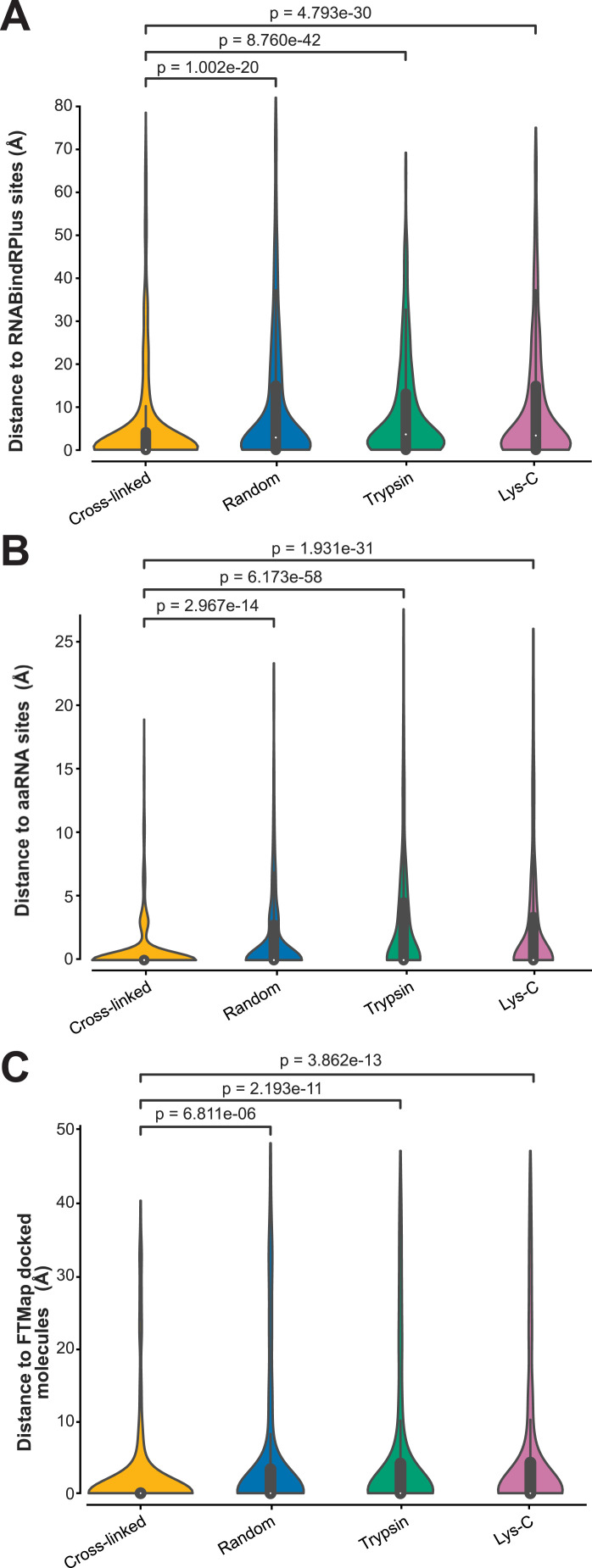

References
-
- Akiba T, Sano S, Yanase T, Ohta T, Koyama M (2019) Optuna: A next-generation hyperparameter optimization framework. arXiv. 10.48550/arXiv.1907.10902 (Preprint posted July 25, 2019). - DOI
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources