

Harnessing DNA replication stress to target RBM10 deficiency in lung adenocarcinoma
- PMID: 39080280
- PMCID: PMC11289143
- DOI: 10.1038/s41467-024-50882-0
Harnessing DNA replication stress to target RBM10 deficiency in lung adenocarcinoma
Abstract
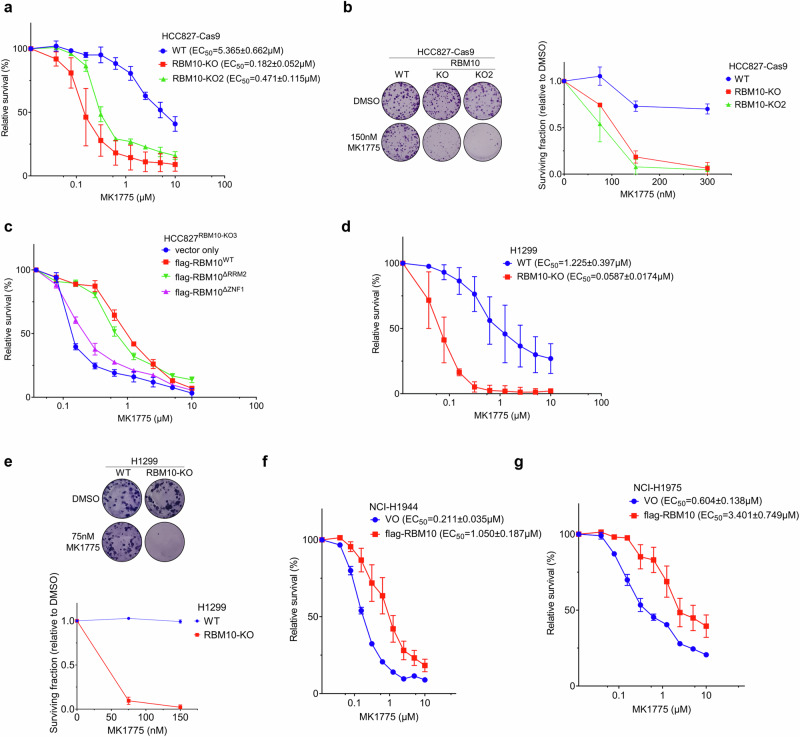
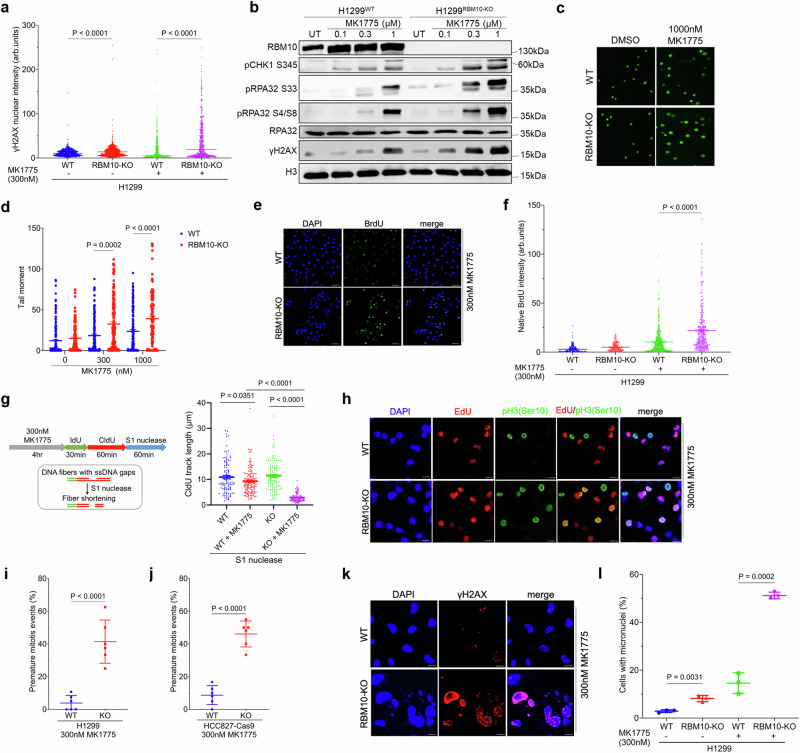
The splicing factor RNA-binding motif protein 10 (RBM10) is frequently mutated in lung adenocarcinoma (LUAD) (9-25%). Most RBM10 cancer mutations are loss-of-function, correlating with increased tumorigenesis and limiting the efficacy of current LUAD targeted therapies. Remarkably, therapeutic strategies leveraging RBM10 deficiency remain unexplored. Here, we conduct a CRISPR-Cas9 synthetic lethality (SL) screen and identify ~60 RBM10 SL genes, including WEE1 kinase. WEE1 inhibition sensitizes RBM10-deficient LUAD cells in-vitro and in-vivo. Mechanistically, we identify a splicing-independent role of RBM10 in regulating DNA replication fork progression and replication stress response, which underpins RBM10-WEE1 SL. Additionally, RBM10 interacts with active DNA replication forks, relying on DNA Primase Subunit 1 (PRIM1) that synthesizes Okazaki RNA primers. Functionally, we demonstrate that RBM10 serves as an anchor for recruiting Histone Deacetylase 1 (HDAC1) to facilitate H4K16 deacetylation and R-loop homeostasis to maintain replication fork stability. Collectively, our data reveal a role of RBM10 in fine-tuning DNA replication and provide therapeutic arsenal for targeting RBM10-deficient tumors.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





Similar articles
-
RNA-binding motif protein 10 represses tumor progression through the Wnt/β- catenin pathway in lung adenocarcinoma.Int J Biol Sci. 2022 Jan 1;18(1):124-139. doi: 10.7150/ijbs.63598. eCollection 2022. Int J Biol Sci. 2022. PMID: 34975322 Free PMC article.
-
RBM10 deficiency promotes brain metastasis by modulating sphingolipid metabolism in a BBB model of EGFR mutant lung adenocarcinoma.J Exp Clin Cancer Res. 2025 Mar 11;44(1):95. doi: 10.1186/s13046-025-03347-1. J Exp Clin Cancer Res. 2025. PMID: 40069781 Free PMC article.
-
RNA binding motif protein 10 suppresses lung cancer progression by controlling alternative splicing of eukaryotic translation initiation factor 4H.EBioMedicine. 2020 Nov;61:103067. doi: 10.1016/j.ebiom.2020.103067. Epub 2020 Oct 23. EBioMedicine. 2020. PMID: 33130397 Free PMC article.
-
RBM10, a New Regulator of p53.Cells. 2020 Sep 16;9(9):2107. doi: 10.3390/cells9092107. Cells. 2020. PMID: 32947864 Free PMC article. Review.
-
RBM10: Structure, functions, and associated diseases.Gene. 2021 May 30;783:145463. doi: 10.1016/j.gene.2021.145463. Epub 2021 Jan 28. Gene. 2021. PMID: 33515724 Free PMC article. Review.
Cited by
-
Immunotherapy in biliary tract cancer: reshaping the tumour microenvironment and advancing precision combination strategies.Front Immunol. 2025 Aug 8;16:1651769. doi: 10.3389/fimmu.2025.1651769. eCollection 2025. Front Immunol. 2025. PMID: 40861435 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
