

Neural network extrapolation to distant regions of the protein fitness landscape
- PMID: 39080282
- PMCID: PMC11289474
- DOI: 10.1038/s41467-024-50712-3
Neural network extrapolation to distant regions of the protein fitness landscape
Abstract
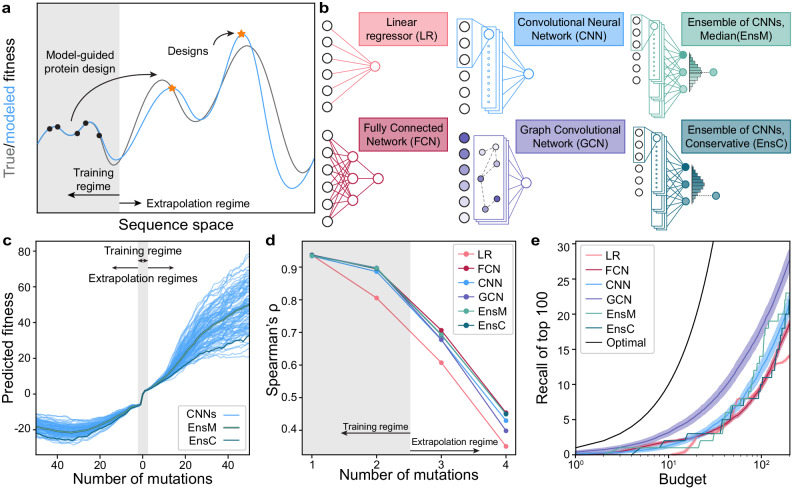
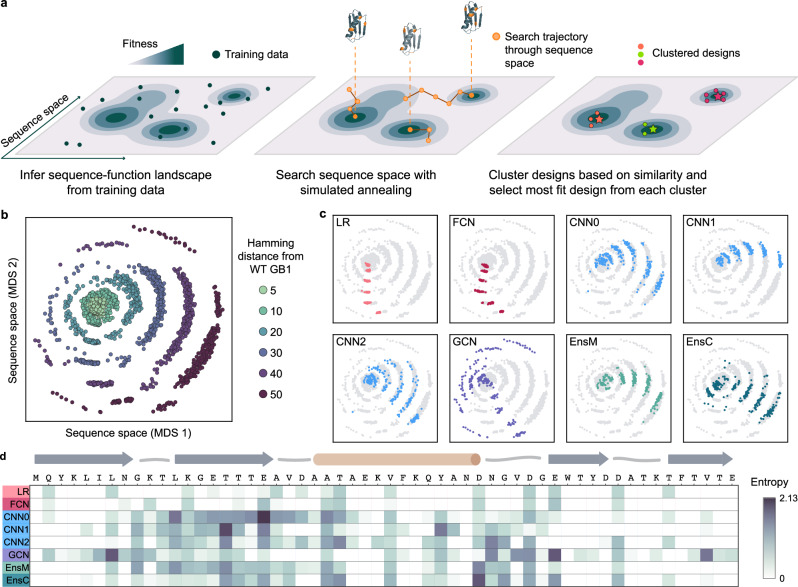
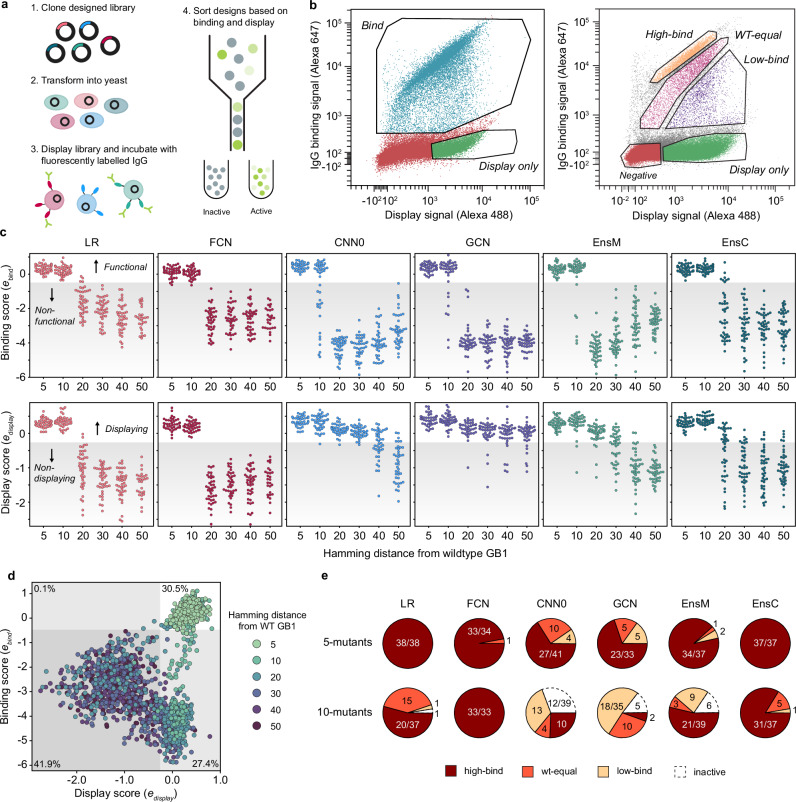
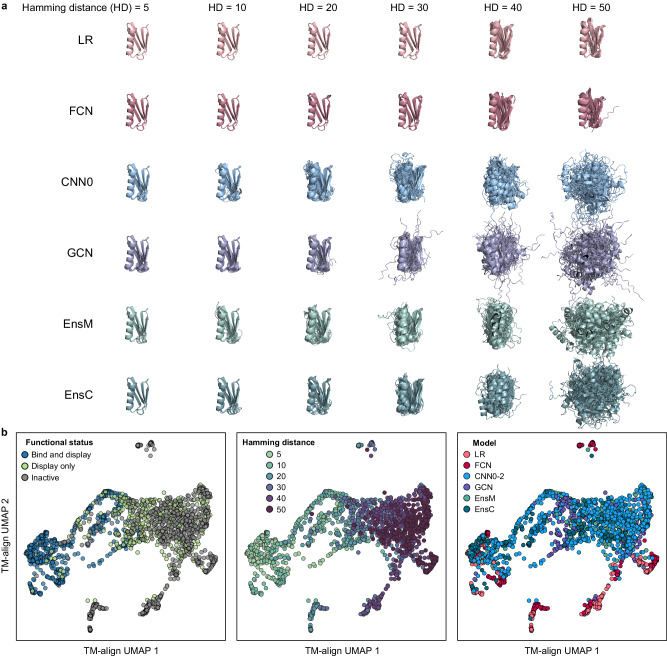
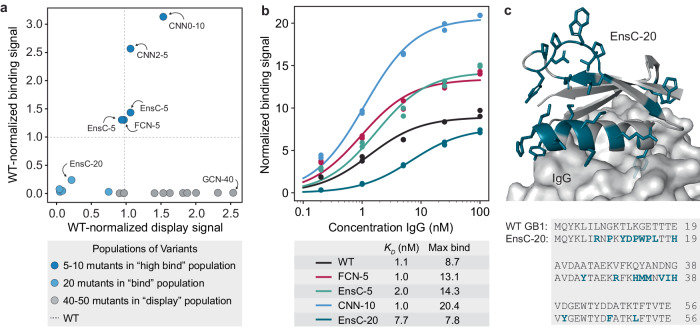
Machine learning (ML) has transformed protein engineering by constructing models of the underlying sequence-function landscape to accelerate the discovery of new biomolecules. ML-guided protein design requires models, trained on local sequence-function information, to accurately predict distant fitness peaks. In this work, we evaluate neural networks' capacity to extrapolate beyond their training data. We perform model-guided design using a panel of neural network architectures trained on protein G (GB1)-Immunoglobulin G (IgG) binding data and experimentally test thousands of GB1 designs to systematically evaluate the models' extrapolation. We find each model architecture infers markedly different landscapes from the same data, which give rise to unique design preferences. We find simpler models excel in local extrapolation to design high fitness proteins, while more sophisticated convolutional models can venture deep into sequence space to design proteins that fold but are no longer functional. We also find that implementing a simple ensemble of convolutional neural networks enables robust design of high-performing variants in the local landscape. Our findings highlight how each architecture's inductive biases prime them to learn different aspects of the protein fitness landscape and how a simple ensembling approach makes protein engineering more robust.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
Neural network extrapolation to distant regions of the protein fitness landscape.bioRxiv [Preprint]. 2023 Nov 9:2023.11.08.566287. doi: 10.1101/2023.11.08.566287. bioRxiv. 2023. Update in: Nat Commun. 2024 Jul 30;15(1):6405. doi: 10.1038/s41467-024-50712-3. PMID: 37987009 Free PMC article. Updated. Preprint.
Similar articles
-
Neural network extrapolation to distant regions of the protein fitness landscape.bioRxiv [Preprint]. 2023 Nov 9:2023.11.08.566287. doi: 10.1101/2023.11.08.566287. bioRxiv. 2023. Update in: Nat Commun. 2024 Jul 30;15(1):6405. doi: 10.1038/s41467-024-50712-3. PMID: 37987009 Free PMC article. Updated. Preprint.
-
Neural networks to learn protein sequence-function relationships from deep mutational scanning data.Proc Natl Acad Sci U S A. 2021 Nov 30;118(48):e2104878118. doi: 10.1073/pnas.2104878118. Proc Natl Acad Sci U S A. 2021. PMID: 34815338 Free PMC article.
-
Genotype sampling for deep-learning assisted experimental mapping of a combinatorially complete fitness landscape.Bioinformatics. 2024 May 2;40(5):btae317. doi: 10.1093/bioinformatics/btae317. Bioinformatics. 2024. PMID: 38745436 Free PMC article.
-
Data Integration Using Advances in Machine Learning in Drug Discovery and Molecular Biology.Methods Mol Biol. 2021;2190:167-184. doi: 10.1007/978-1-0716-0826-5_7. Methods Mol Biol. 2021. PMID: 32804365 Review.
-
Machine learning to navigate fitness landscapes for protein engineering.Curr Opin Biotechnol. 2022 Jun;75:102713. doi: 10.1016/j.copbio.2022.102713. Epub 2022 Apr 9. Curr Opin Biotechnol. 2022. PMID: 35413604 Free PMC article. Review.
Cited by
-
Investigating the determinants of performance in machine learning for protein fitness prediction.Protein Sci. 2025 Aug;34(8):e70235. doi: 10.1002/pro.70235. Protein Sci. 2025. PMID: 40689706 Free PMC article.
-
Machine learning in molecular biophysics: Protein allostery, multi-level free energy simulations, and lipid phase transitions.Biophys Rev (Melville). 2025 Feb 12;6(1):011305. doi: 10.1063/5.0248589. eCollection 2025 Mar. Biophys Rev (Melville). 2025. PMID: 39957913 Review.
-
Opportunities and Challenges for Machine Learning-Assisted Enzyme Engineering.ACS Cent Sci. 2024 Feb 5;10(2):226-241. doi: 10.1021/acscentsci.3c01275. eCollection 2024 Feb 28. ACS Cent Sci. 2024. PMID: 38435522 Free PMC article. Review.
-
Development of the autonomous lab system to support biotechnology research.Sci Rep. 2025 Feb 24;15(1):6648. doi: 10.1038/s41598-025-89069-y. Sci Rep. 2025. PMID: 39994271 Free PMC article.
-
Designing diverse and high-performance proteins with a large language model in the loop.PLoS Comput Biol. 2025 Jun 5;21(6):e1013119. doi: 10.1371/journal.pcbi.1013119. eCollection 2025 Jun. PLoS Comput Biol. 2025. PMID: 40471987 Free PMC article.
References
MeSH terms
Substances
Grants and funding
- R35 GM119854/GM/NIGMS NIH HHS/United States
- P30 CA014520/CA/NCI NIH HHS/United States
- T32 HG002760/HG/NHGRI NIH HHS/United States
- S10 OD018202/OD/NIH HHS/United States
- 5R35GM119854/U.S. Department of Health & Human Services | NIH | Center for Information Technology (Center for Information Technology, National Institutes of Health)
LinkOut - more resources
Full Text Sources
