

mTORC2-driven chromatin cGAS mediates chemoresistance through epigenetic reprogramming in colorectal cancer
- PMID: 39080411
- PMCID: PMC11392818
- DOI: 10.1038/s41556-024-01473-0
mTORC2-driven chromatin cGAS mediates chemoresistance through epigenetic reprogramming in colorectal cancer
Abstract
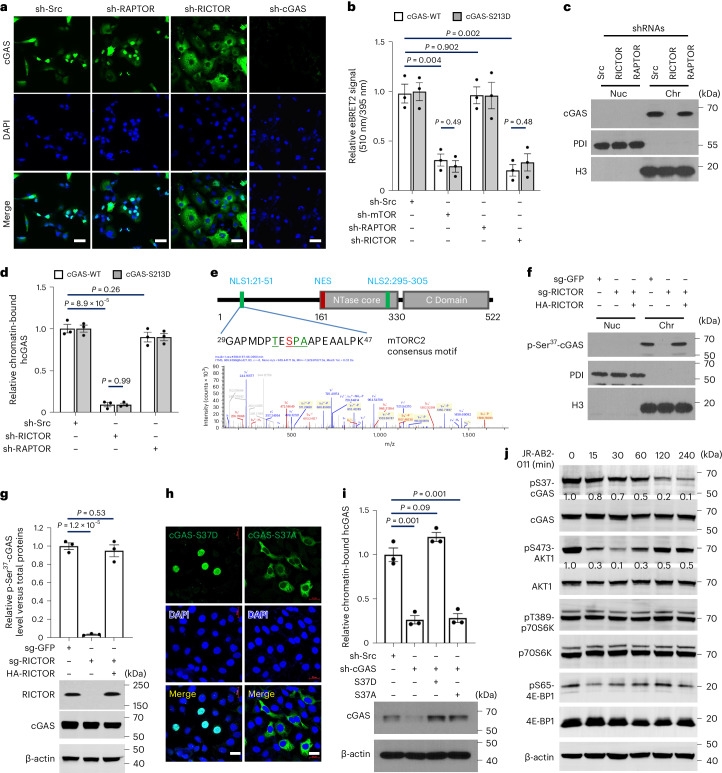
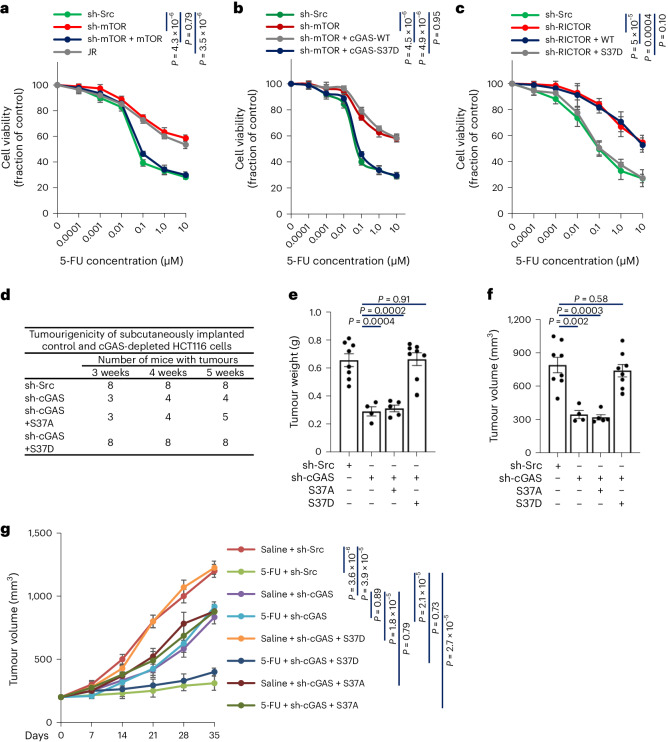
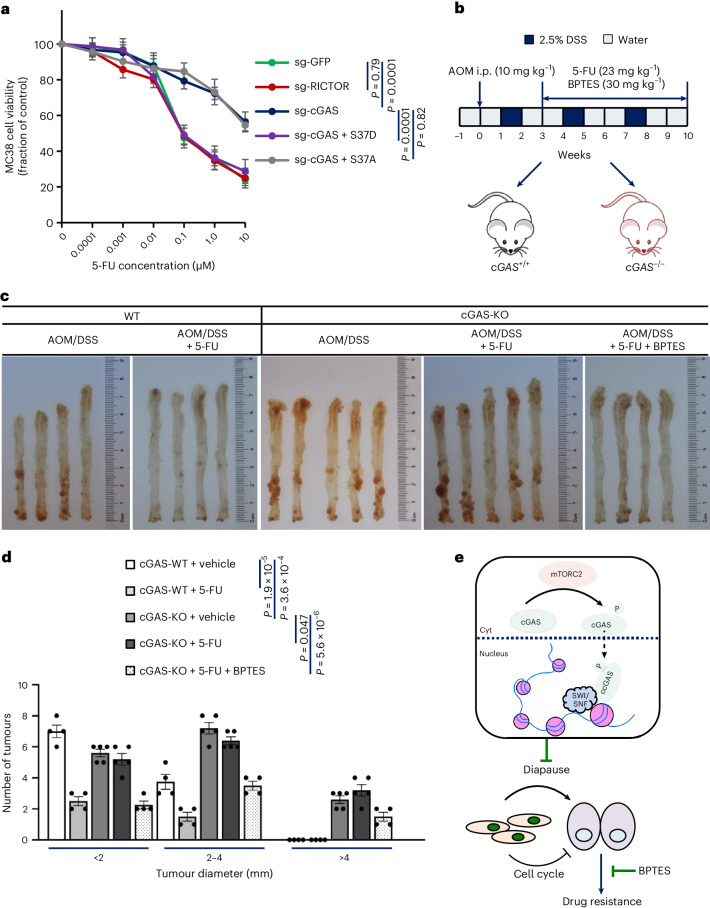
Cyclic GMP-AMP synthase (cGAS), a cytosolic DNA sensor that initiates a STING-dependent innate immune response, binds tightly to chromatin, where its catalytic activity is inhibited; however, mechanisms underlying cGAS recruitment to chromatin and functions of chromatin-bound cGAS (ccGAS) remain unclear. Here we show that mTORC2-mediated phosphorylation of human cGAS serine 37 promotes its chromatin localization in colorectal cancer cells, regulating cell growth and drug resistance independently of STING. We discovered that ccGAS recruits the SWI/SNF complex at specific chromatin regions, modifying expression of genes linked to glutaminolysis and DNA replication. Although ccGAS depletion inhibited cell growth, it induced chemoresistance to fluorouracil treatment in vitro and in vivo. Moreover, blocking kidney-type glutaminase, a downstream ccGAS target, overcame chemoresistance caused by ccGAS loss. Thus, ccGAS coordinates colorectal cancer plasticity and acquired chemoresistance through epigenetic patterning. Targeting both mTORC2-ccGAS and glutaminase provides a promising strategy to eliminate quiescent resistant cancer cells.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures














References
-
- Chen, C. & Xu, P. Cellular functions of cGAS-STING signaling. Trends Cell Biol.10.1016/j.tcb.2022.11.001 (2022). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 2024A1515030060/Natural Science Foundation of Guangdong Province (Guangdong Natural Science Foundation)
- 81602422/National Natural Science Foundation of China (National Science Foundation of China)
- 81903094/National Natural Science Foundation of China (National Science Foundation of China)
- 2023J030/Natural Science Foundation of Ningbo (Ningbo Natural Science Foundation)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
