

A Novel Monoallelic ALG5 Variant Causing Late-Onset ADPKD and Tubulointerstitial Fibrosis
- PMID: 39081747
- PMCID: PMC11284371
- DOI: 10.1016/j.ekir.2024.04.031
A Novel Monoallelic ALG5 Variant Causing Late-Onset ADPKD and Tubulointerstitial Fibrosis
Abstract
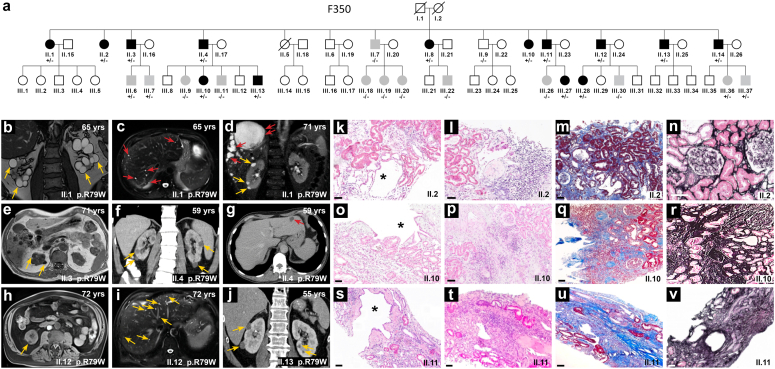
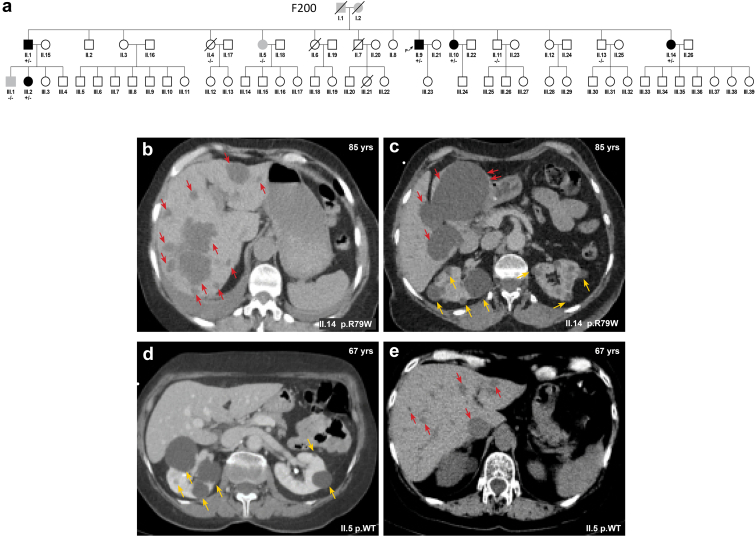
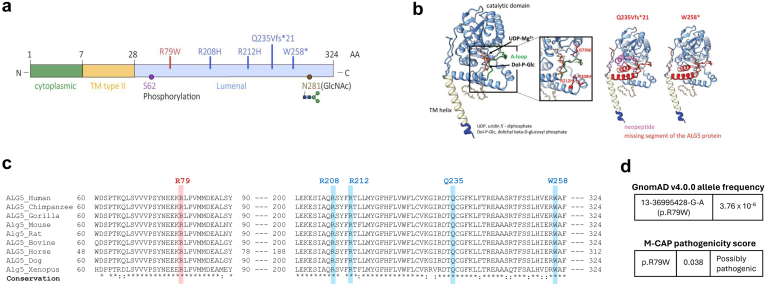
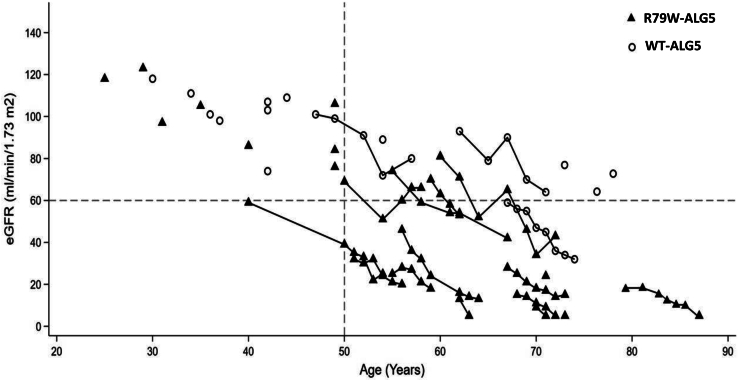
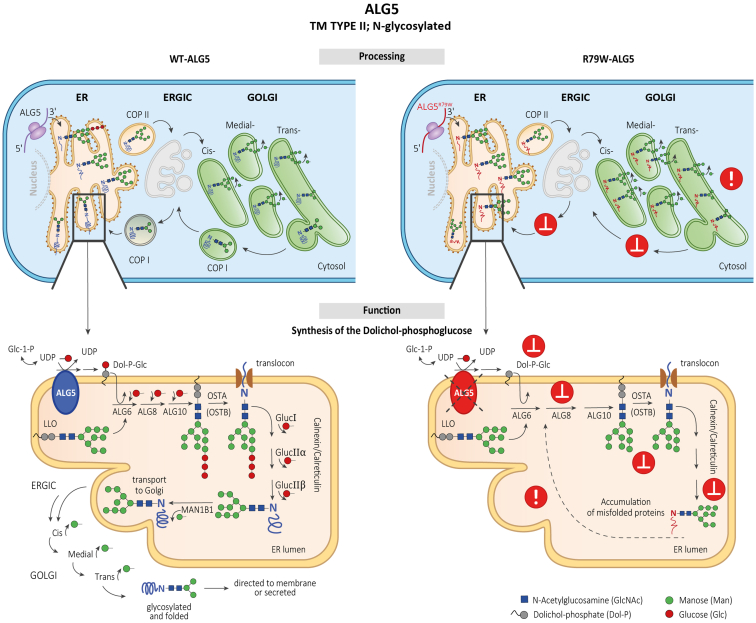
Introduction: Monoallelic variants in the ALG5 gene encoding asparagine-linked glycosylation protein 5 homolog (ALG5) have been recently shown to disrupt polycystin-1 (PC1) maturation and trafficking via underglycosylation, causing an autosomal dominant polycystic kidney disease-like (ADPKD-like) phenotype and interstitial fibrosis. In this report, we present clinical, genetic, histopathologic, and protein structure and functional correlates of a new ALG5 variant, p.R79W, that we identified in 2 distant genetically related Irish families displaying an atypical late-onset ADPKD phenotype combined with tubulointerstitial damage.
Methods: Whole exome and targeted sequencing were used for segregation analysis of available relatives. This was followed by immunohistochemistry examinations of kidney biopsies, and targeted (UMOD, MUC1) and untargeted plasma proteome and N-glycomic studies.
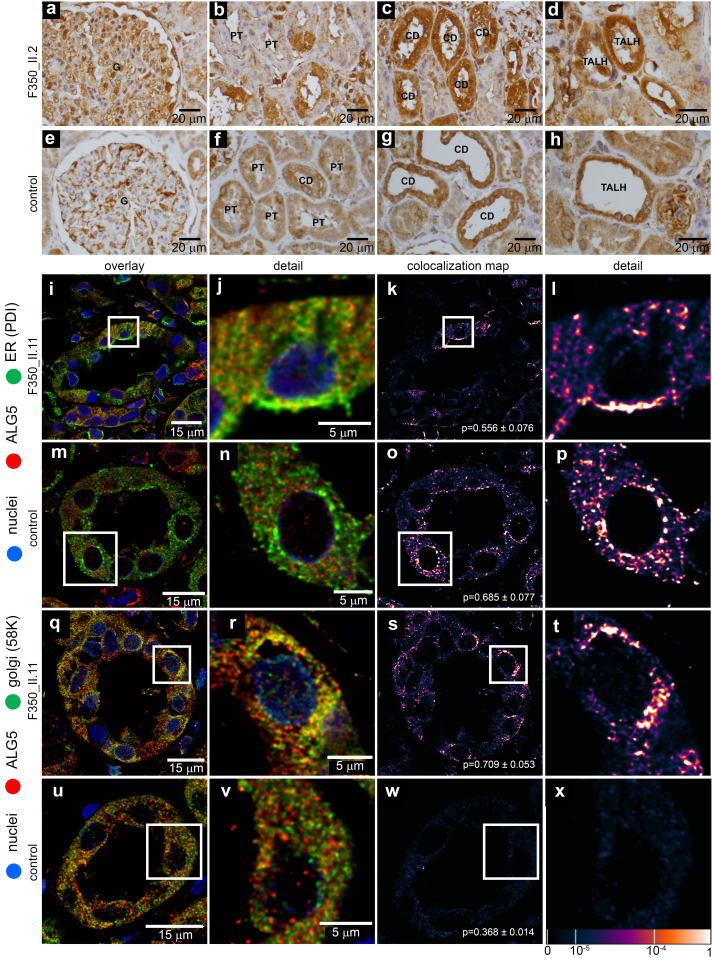
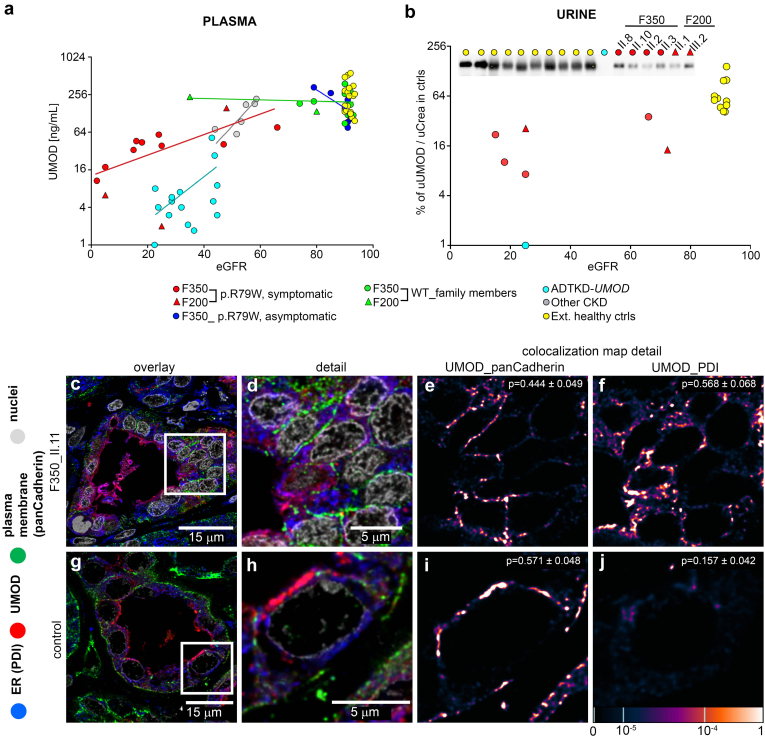
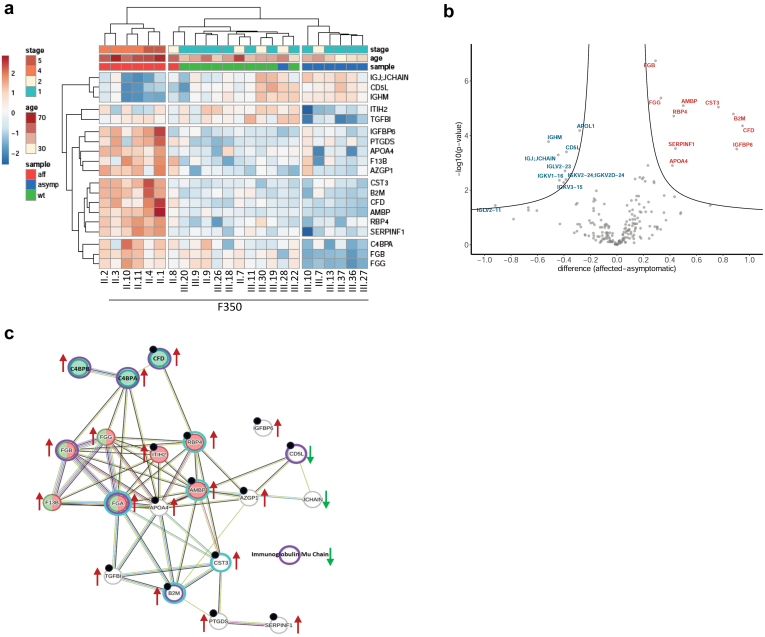
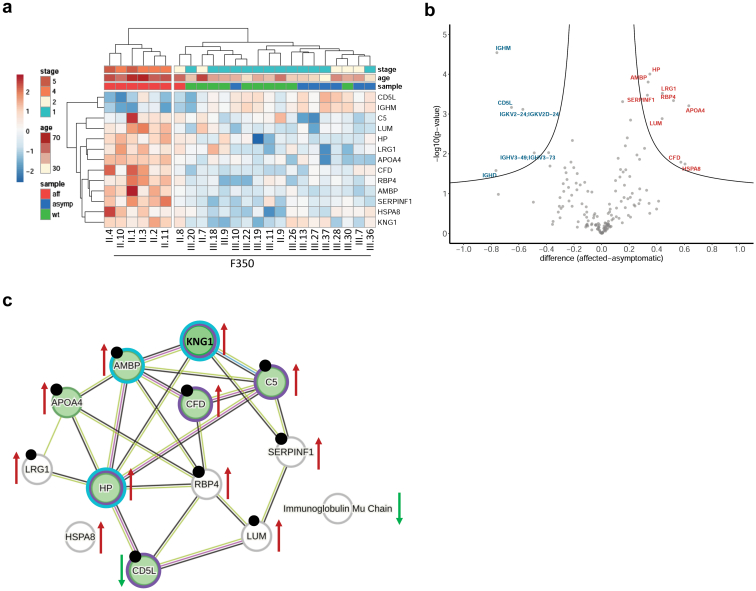
Results: We identified a monoallelic ALG5 variant [GRCh37 (NM_013338.5): g.37569565G>A, c.235C>T; p.R79W] that cosegregates in 23 individuals, of whom 18 were clinically affected. We detected abnormal localization of ALG5 in the Golgi apparatus of renal tubular cells in patients' kidney specimens. Further, we detected the pathological accumulation of uromodulin, an N-glycosylated glycosylphosphatidylinositol (GPI)-anchored protein, in the endoplasmic reticulum (ER), but not mucin-1, an O- and N-glycosylated protein. Biochemical investigation revealed decreased plasma and urinary uromodulin levels in clinically affected individuals. Proteomic and glycoproteomic profiling revealed the dysregulation of chronic kidney disease (CKD)-associated proteins.
Conclusion: ALG5 dysfunction adversely affects maturation and trafficking of N-glycosylated and GPI anchored protein uromodulin, leading to structural and functional changes in the kidney. Our findings confirm ALG5 as a cause of late-onset ADPKD and provide additional insight into the molecular mechanisms of ADPKD-ALG5.
Keywords: ALG5; Golgi apparatus; N-Linked protein glycosylation; UMOD; autosomal dominant tubulo-interstitial kidney disease; autosomal-dominant polycystic kidney disease.
© 2024 International Society of Nephrology. Published by Elsevier Inc.
Figures
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
