

Adsorption-Driven Deformation and Footprints of the RBD Proteins in SARS-CoV-2 Variants on Biological and Inanimate Surfaces
- PMID: 39083670
- PMCID: PMC11323246
- DOI: 10.1021/acs.jcim.4c00460
Adsorption-Driven Deformation and Footprints of the RBD Proteins in SARS-CoV-2 Variants on Biological and Inanimate Surfaces
Abstract
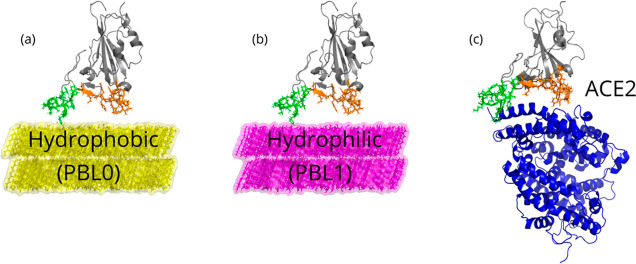
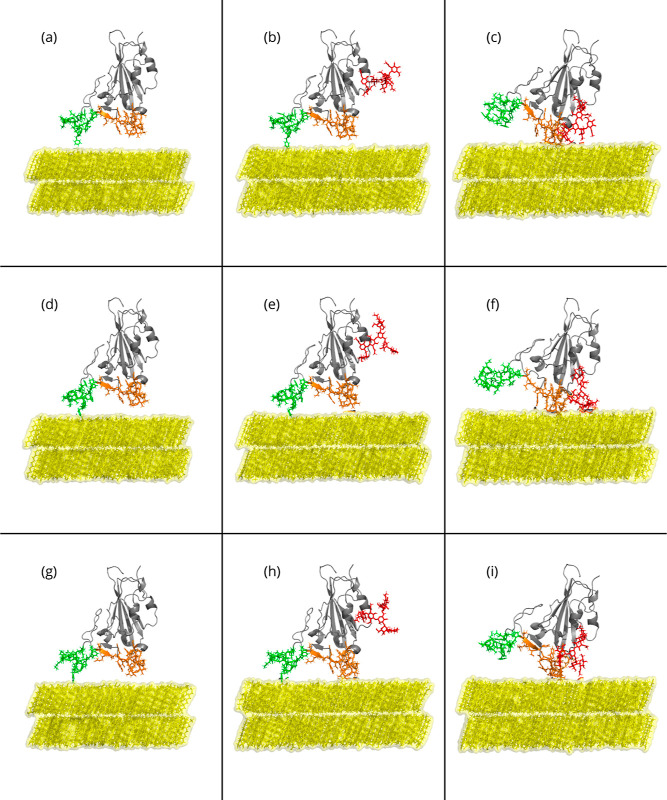
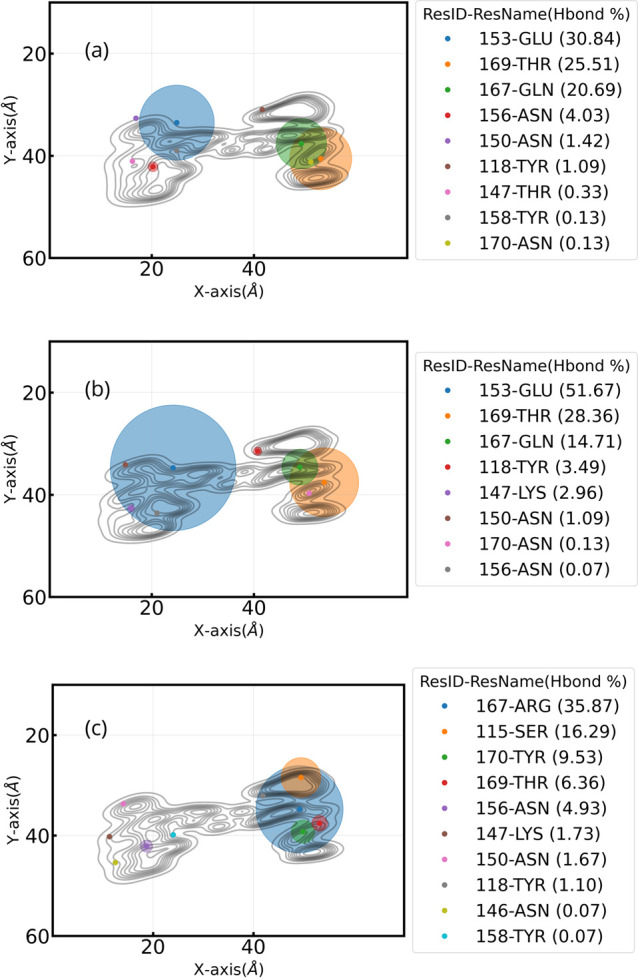
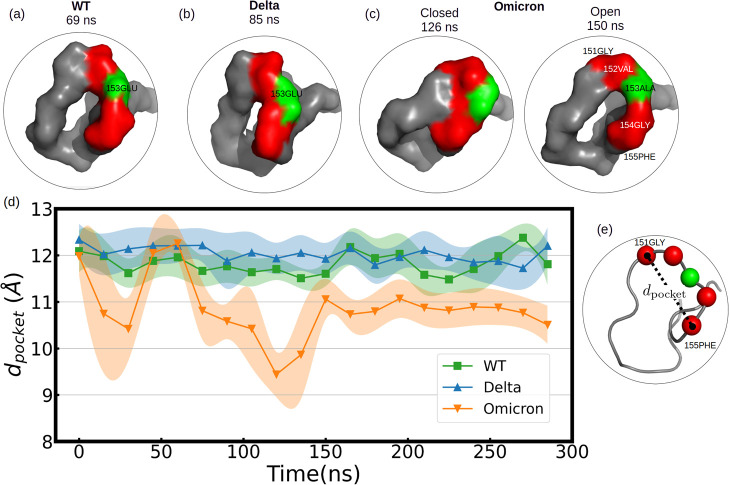
Respiratory viruses, carried through airborne microdroplets, frequently adhere to surfaces, including plastics and metals. However, our understanding of the interactions between viruses and materials remains limited, particularly in scenarios involving polarizable surfaces. Here, we investigate the role of the receptor-binding domain (RBD) of the spike protein mutations on the adsorption of SARS-CoV-2 to hydrophobic and hydrophilic surfaces employing molecular simulations. To contextualize our findings, we contrast the interactions on inanimate surfaces with those on native biological interfaces, specifically the angiotensin-converting enzyme 2. Notably, we identify a 2-fold increase in structural deformations for the protein's receptor binding motif (RBM) onto inanimate surfaces, indicative of enhanced shock-absorbing mechanisms. Furthermore, the distribution of adsorbed amino acids (landing footprints) on the inanimate surface reveals a distinct regional asymmetry relative to the biological interface, with roughly half of the adsorbed amino acids arranged in opposite sites. In spite of the H-bonds formed at the hydrophilic substrate, the simulations consistently show a higher number of contacts and interfacial area with the hydrophobic surface, where the wild-type RBD adsorbs more strongly than the Delta or Omicron RBDs. In contrast, the adsorption of Delta and Omicron to hydrophilic surfaces was characterized by a distinctive hopping-pattern. The novel shock-absorbing mechanisms identified in the virus adsorption on inanimate surfaces show the embedded high-deformation capacity of the RBD without losing its secondary structure, which could lead to current experimental strategies in the design of virucidal surfaces.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
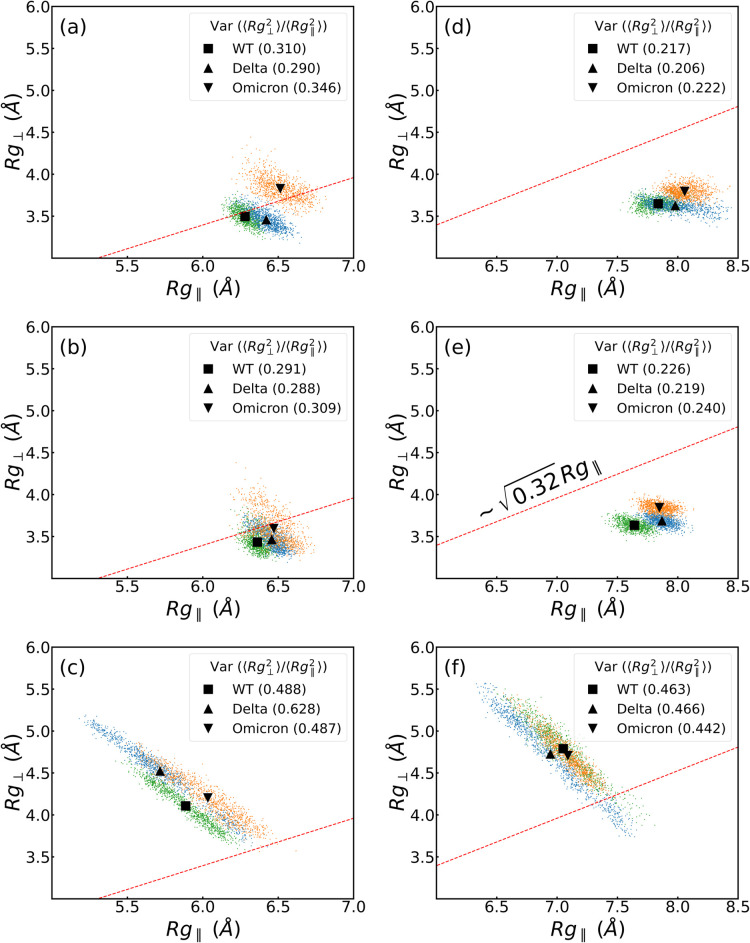
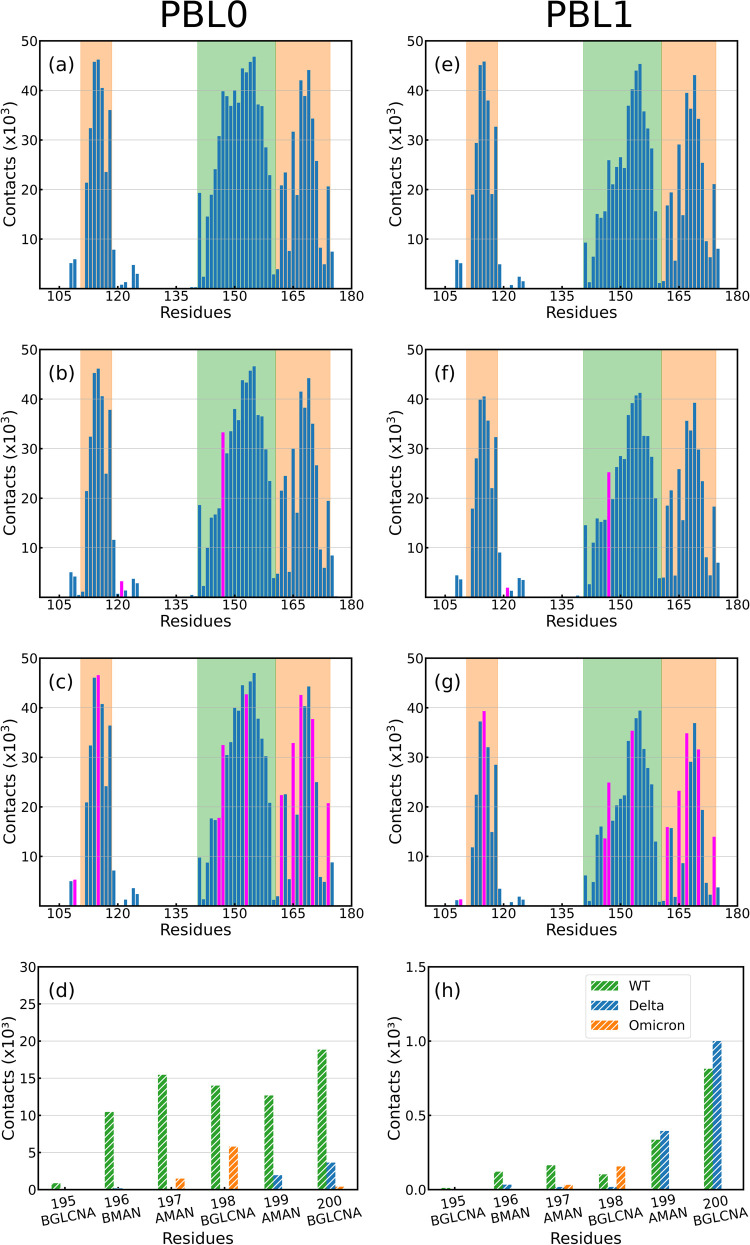
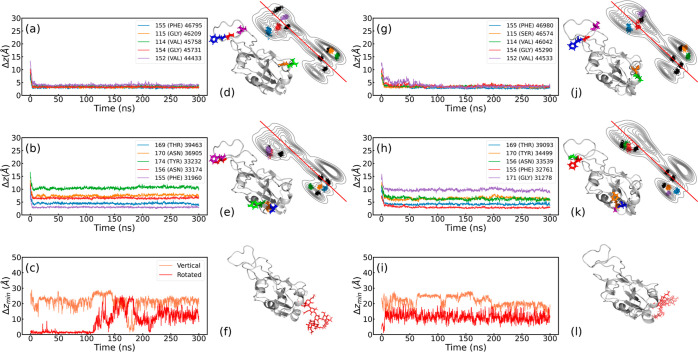
 ratio is shown for each case.
ratio is shown for each case.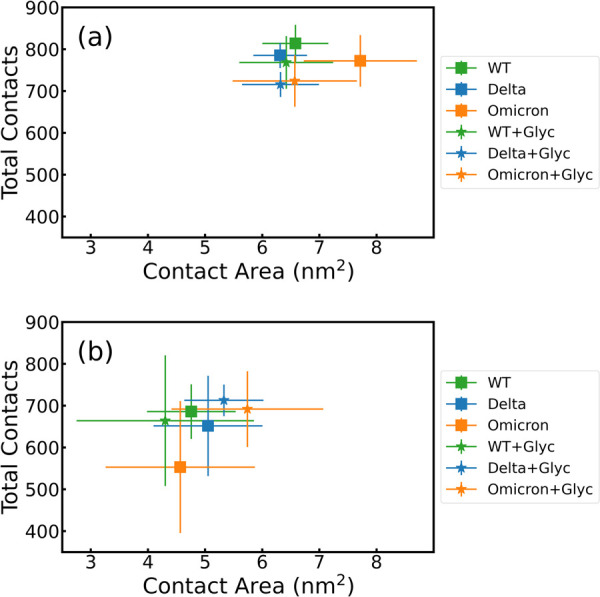

Similar articles
-
V367F Mutation in SARS-CoV-2 Spike RBD Emerging during the Early Transmission Phase Enhances Viral Infectivity through Increased Human ACE2 Receptor Binding Affinity.J Virol. 2021 Jul 26;95(16):e0061721. doi: 10.1128/JVI.00617-21. Epub 2021 Jul 26. J Virol. 2021. PMID: 34105996 Free PMC article.
-
Omicron and Delta variant of SARS-CoV-2: A comparative computational study of spike protein.J Med Virol. 2022 Apr;94(4):1641-1649. doi: 10.1002/jmv.27526. Epub 2021 Dec 27. J Med Virol. 2022. PMID: 34914115
-
Structural basis and analysis of hamster ACE2 binding to different SARS-CoV-2 spike RBDs.J Virol. 2024 Mar 19;98(3):e0115723. doi: 10.1128/jvi.01157-23. Epub 2024 Feb 2. J Virol. 2024. PMID: 38305152 Free PMC article.
-
Molecular Dynamics Studies on the Structural Characteristics for the Stability Prediction of SARS-CoV-2.Int J Mol Sci. 2021 Aug 13;22(16):8714. doi: 10.3390/ijms22168714. Int J Mol Sci. 2021. PMID: 34445414 Free PMC article. Review.
-
Structural basis of severe acute respiratory syndrome coronavirus 2 infection.Curr Opin HIV AIDS. 2021 Jan;16(1):74-81. doi: 10.1097/COH.0000000000000658. Curr Opin HIV AIDS. 2021. PMID: 33186231 Review.
Cited by
-
2Danalysis: A toolbox for analysis of lipid membranes and biopolymers in two-dimensional space.bioRxiv [Preprint]. 2025 Mar 2:2025.02.27.640563. doi: 10.1101/2025.02.27.640563. bioRxiv. 2025. Update in: Biophys J. 2025 May 30:S0006-3495(25)00321-2. doi: 10.1016/j.bpj.2025.05.026. PMID: 40060461 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Miscellaneous
