

Identification and Characterization of Chemotherapy-Resistant High-Risk Neuroblastoma Persister Cells
- PMID: 39083807
- PMCID: PMC11609622
- DOI: 10.1158/2159-8290.CD-24-0046
Identification and Characterization of Chemotherapy-Resistant High-Risk Neuroblastoma Persister Cells
Abstract
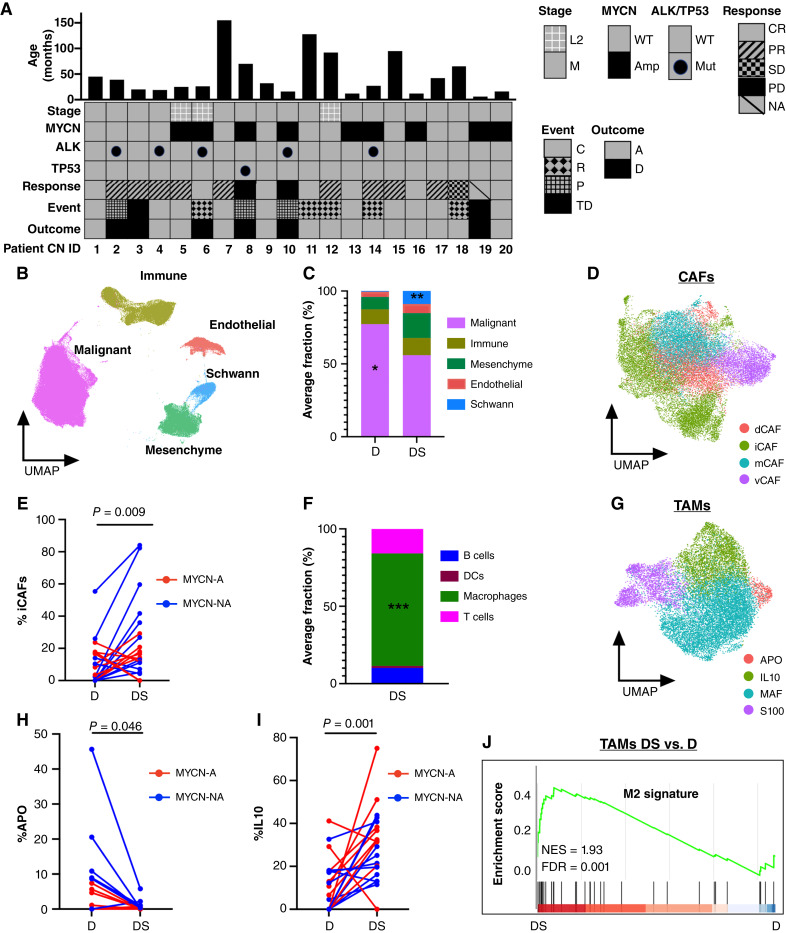
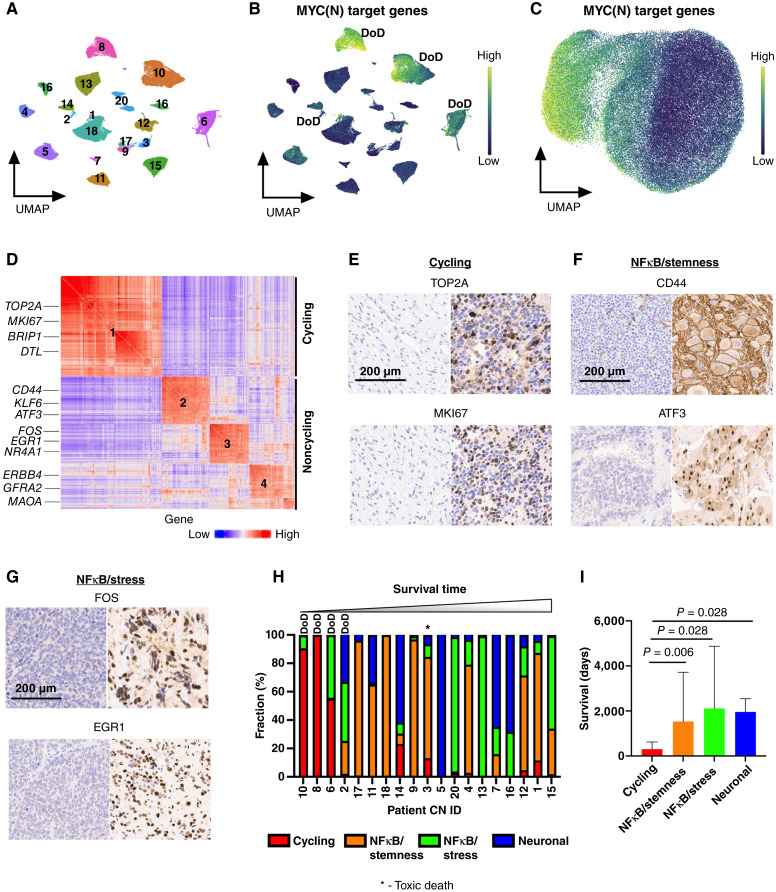
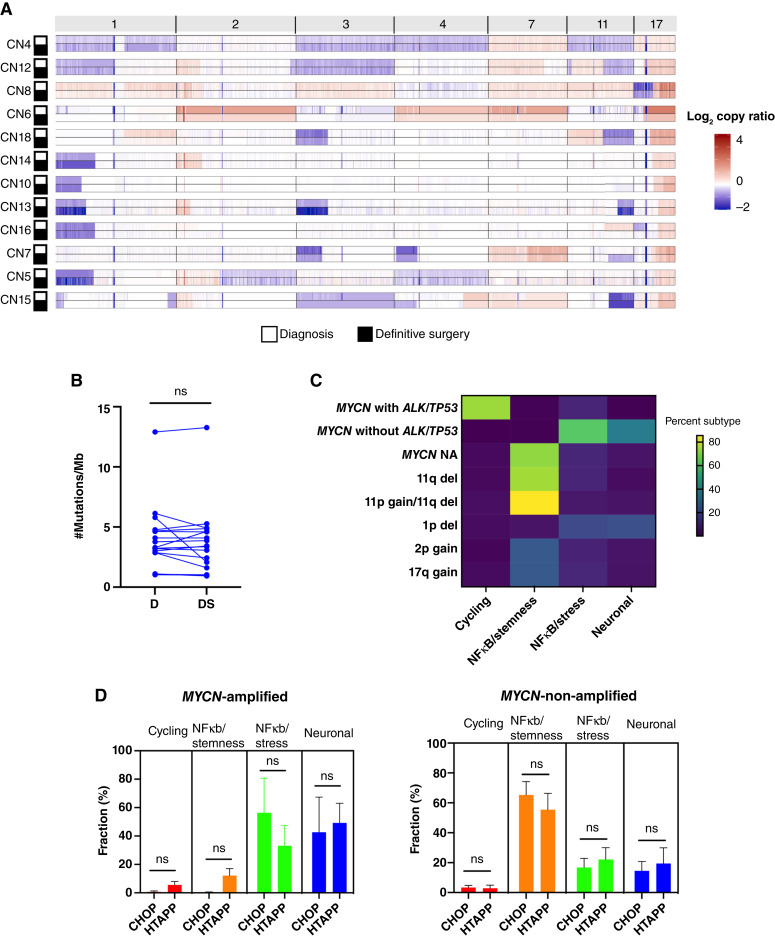
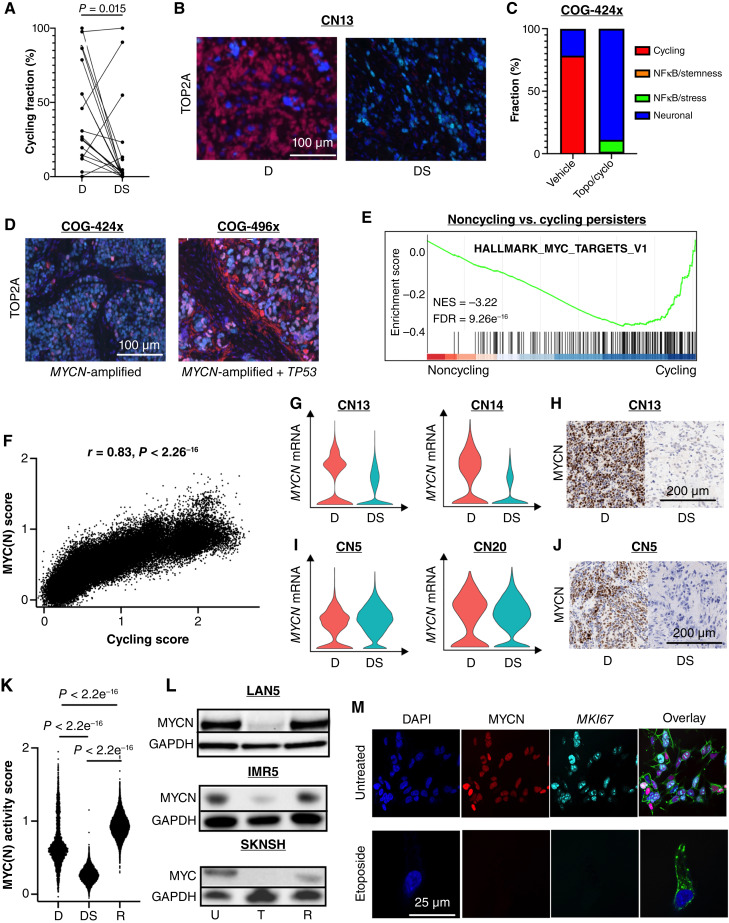
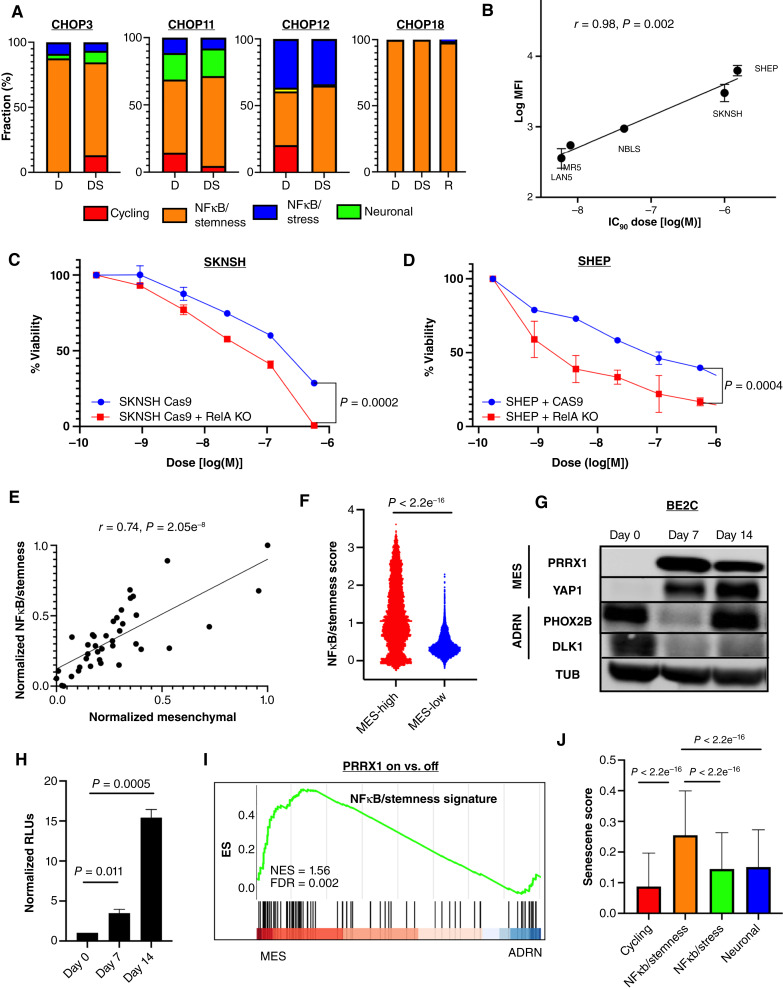
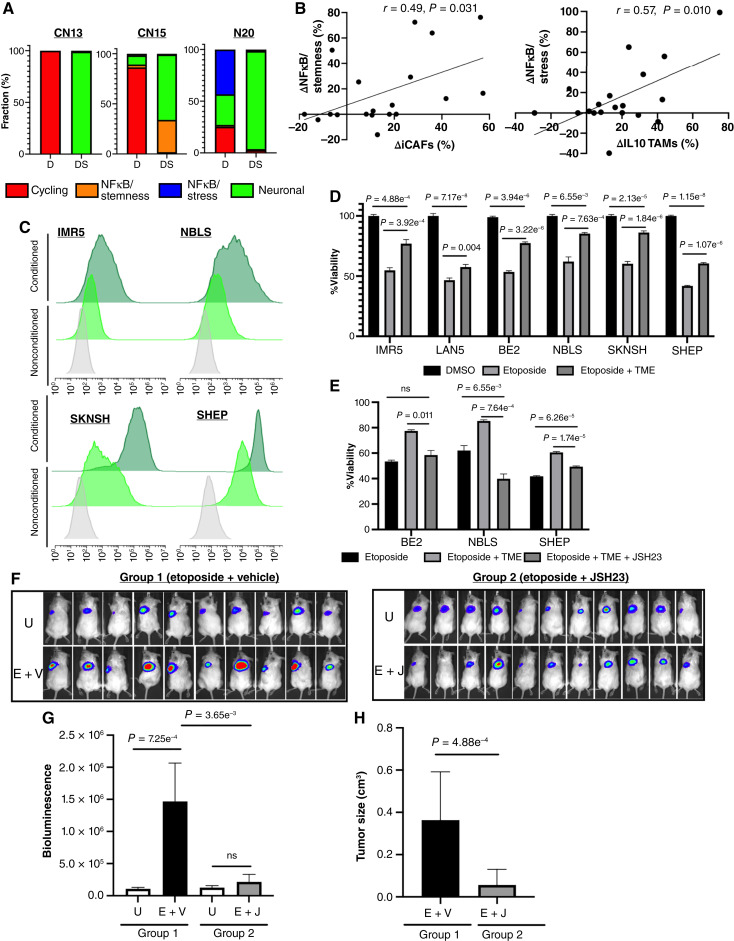
Relapse rates in high-risk neuroblastoma remain exceedingly high. The malignant cells that are responsible for relapse have not been identified, and mechanisms of therapy resistance remain poorly understood. In this study, we used single-nucleus RNA sequencing and bulk whole-genome sequencing to identify and characterize the residual malignant persister cells that survive chemotherapy from a cohort of 20 matched diagnosis and definitive surgery tumor samples from patients treated with high-risk neuroblastoma induction chemotherapy. We show that persister cells share common mechanisms of chemotherapy escape, including suppression of MYC(N) activity and activation of NFκB signaling, and the latter is further enhanced by cell-cell communication between the malignant cells and the tumor microenvironment. Overall, our work dissects the transcriptional landscape of cellular persistence in high-risk neuroblastoma and paves the way to the development of new therapeutic strategies to prevent disease relapse. Significance: Approximately 50% of patients with high-risk neuroblastoma die of relapsed refractory disease. We identified the malignant cells that likely contribute to relapse and discovered key signaling pathways that mediate cellular persistence. Inhibition of these pathways and their downstream effectors is postulated to eliminate persister cells and prevent relapse. See related commentary by Wolf et al., p. 2308.
©2024 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
No disclosures were reported by the authors.
Figures
References
-
- Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. . Neuroblastoma. Nat Rev Dis Primers 2016;2:16078. - PubMed
-
- Norris MD, Bordow SB, Marshall GM, Haber PS, Cohn SL, Haber M. Expression of the gene for multidrug-resistance-associated protein and outcome in patients with neuroblastoma. N Engl J Med 1996;334:231–8. - PubMed
-
- Schramm A, Köster J, Assenov Y, Althoff K, Peifer M, Mahlow E, et al. . Mutational dynamics between primary and relapse neuroblastomas. Nat Genet 2015;47:872–7. - PubMed
-
- Boeva V, Louis-Brennetot C, Peltier A, Durand S, Pierre-Eugène C, Raynal V, et al. . Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat Genet 2017;49:1408–13. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
