

Quantification of escape from X chromosome inactivation with single-cell omics data reveals heterogeneity across cell types and tissues
- PMID: 39084228
- PMCID: PMC11406184
- DOI: 10.1016/j.xgen.2024.100625
Quantification of escape from X chromosome inactivation with single-cell omics data reveals heterogeneity across cell types and tissues
Abstract
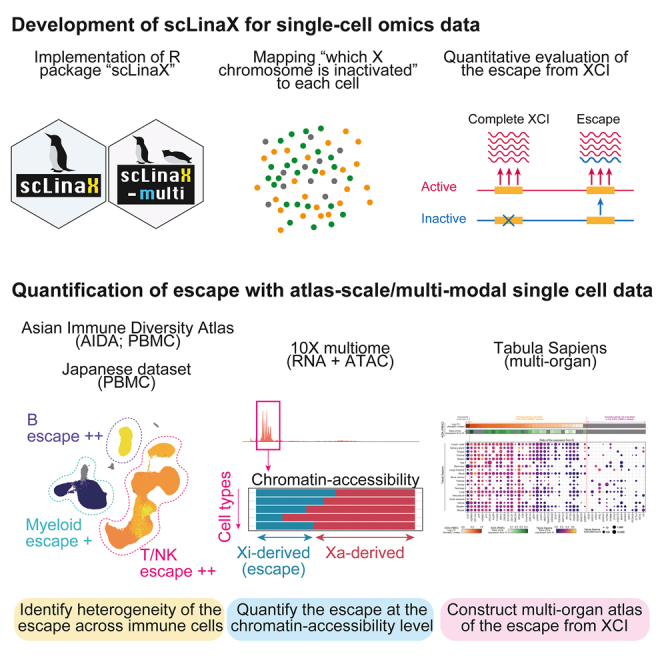
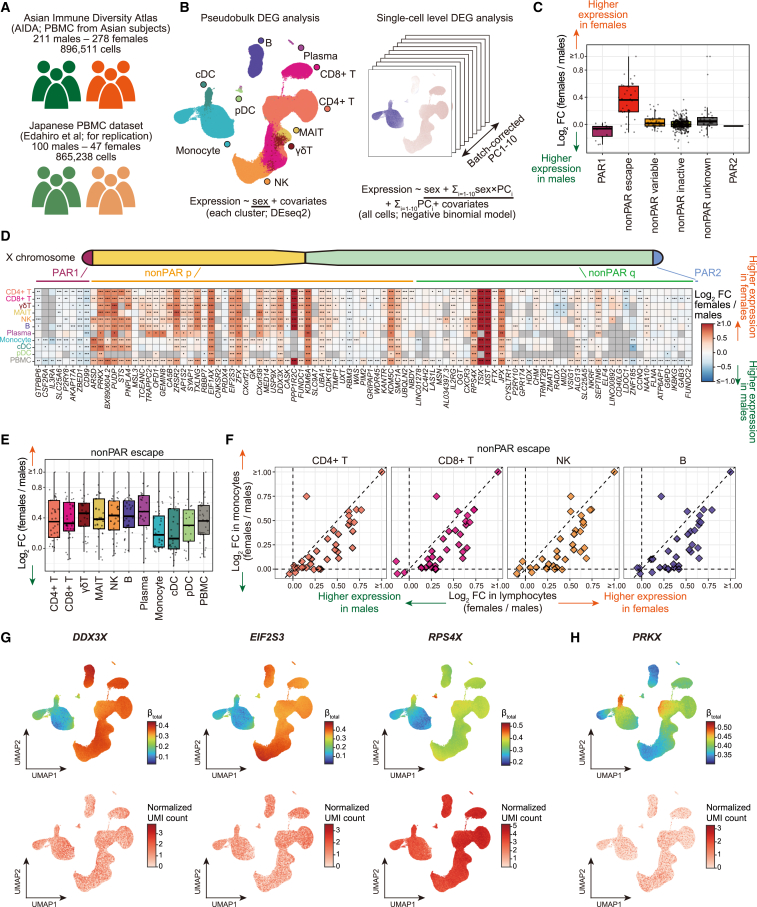
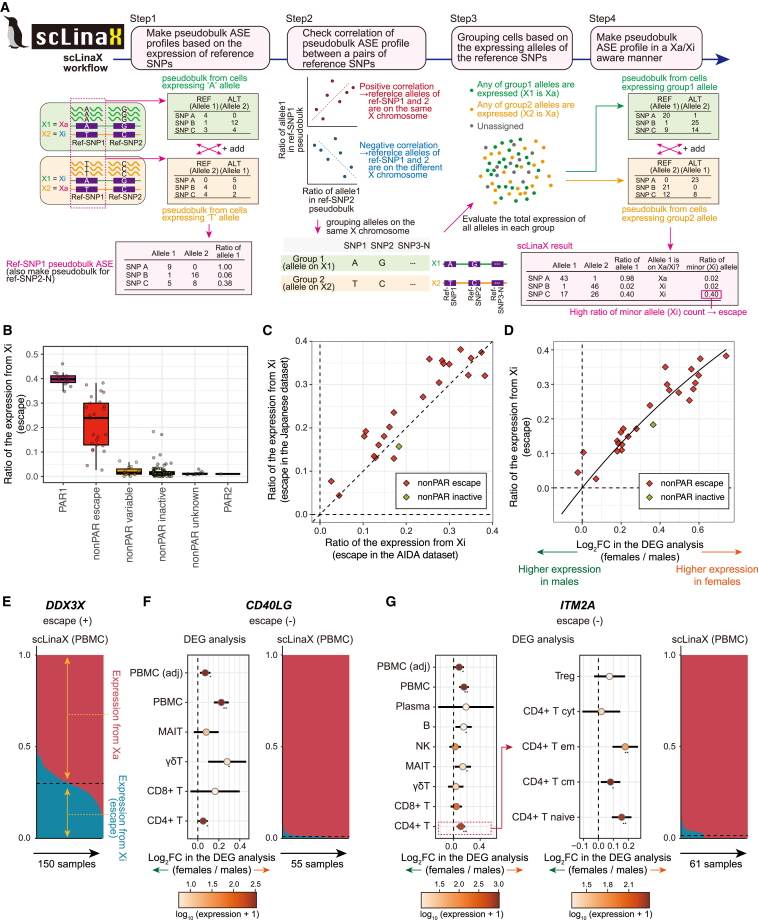
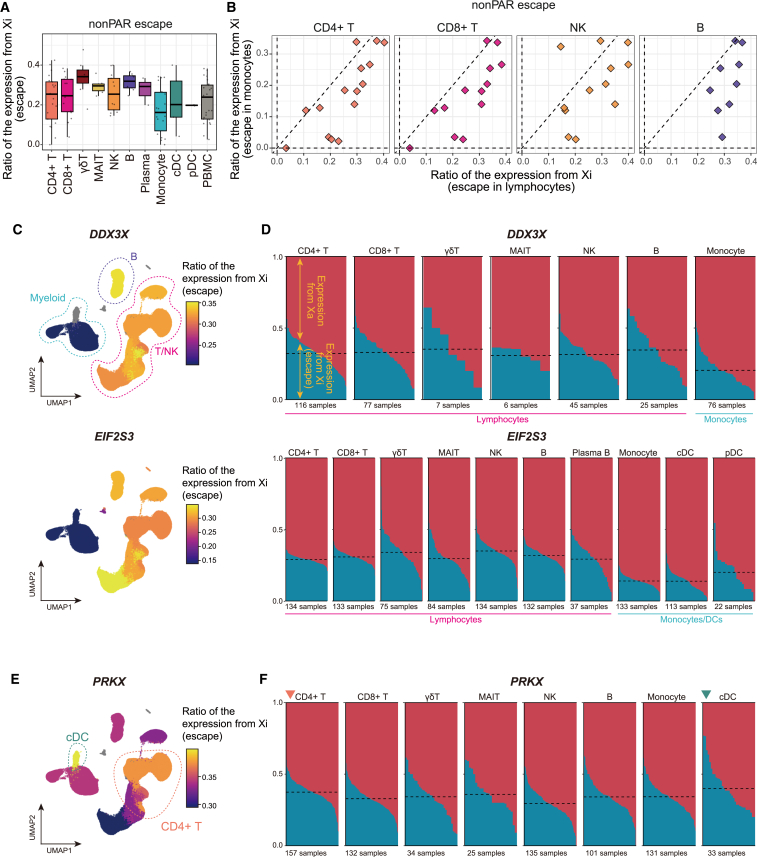
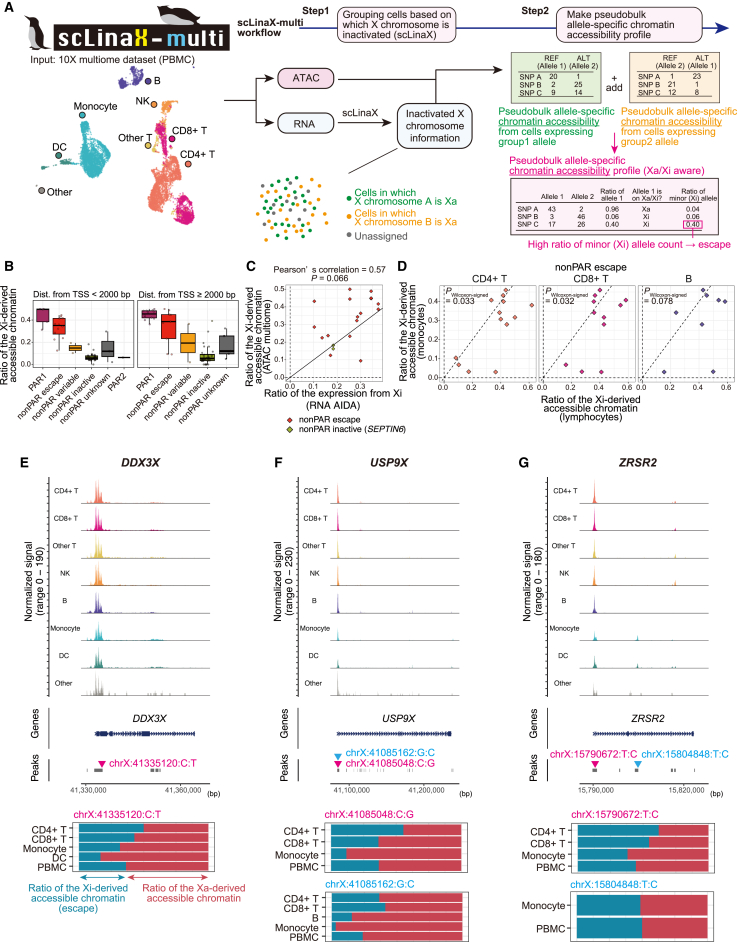
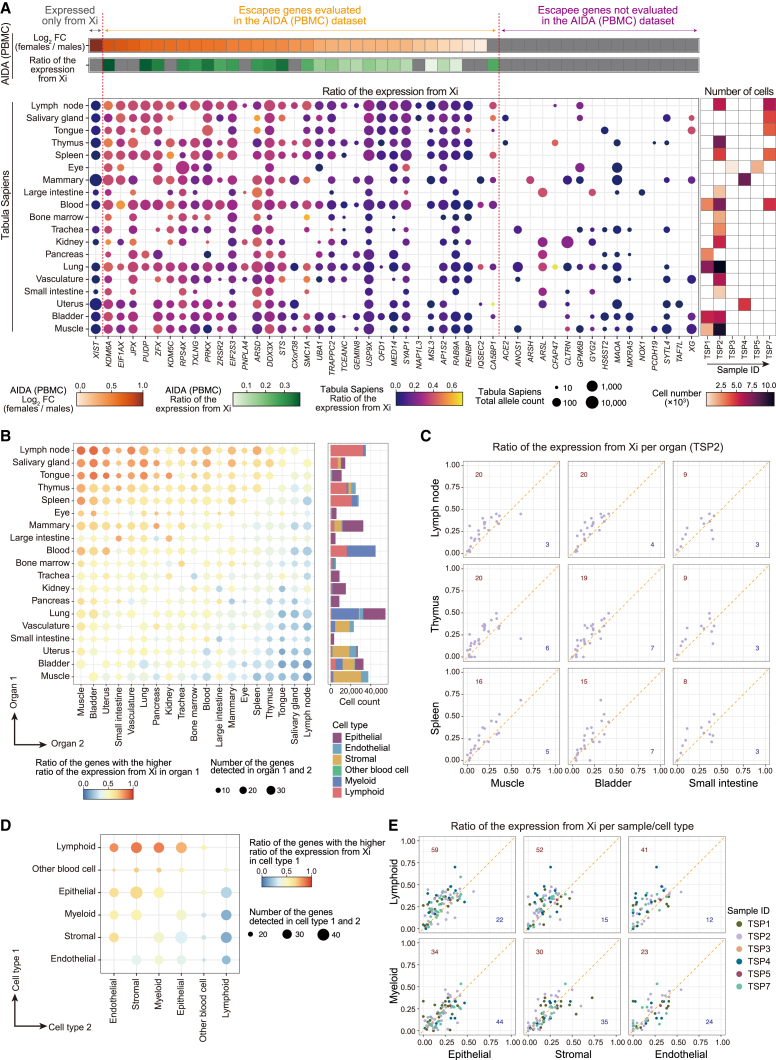
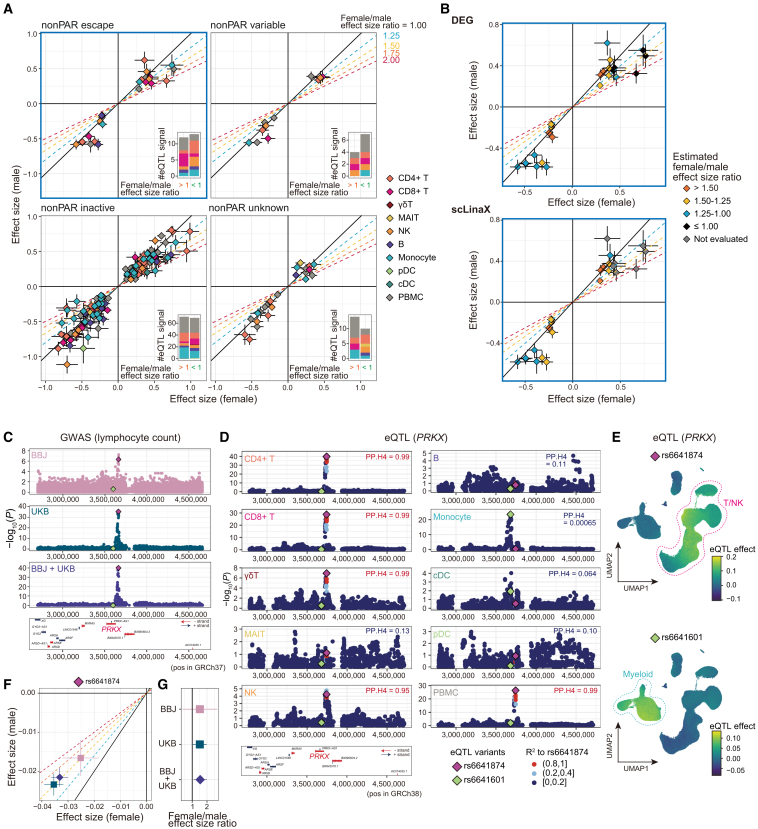
Several X-linked genes escape from X chromosome inactivation (XCI), while differences in escape across cell types and tissues are still poorly characterized. Here, we developed scLinaX for directly quantifying relative gene expression from the inactivated X chromosome with droplet-based single-cell RNA sequencing (scRNA-seq) data. The scLinaX and differentially expressed gene analyses with large-scale blood scRNA-seq datasets consistently identified the stronger escape in lymphocytes than in myeloid cells. An extension of scLinaX to a 10x multiome dataset (scLinaX-multi) suggested a stronger escape in lymphocytes than in myeloid cells at the chromatin-accessibility level. The scLinaX analysis of human multiple-organ scRNA-seq datasets also identified the relatively strong degree of escape from XCI in lymphoid tissues and lymphocytes. Finally, effect size comparisons of genome-wide association studies between sexes suggested the underlying impact of escape on the genotype-phenotype association. Overall, scLinaX and the quantified escape catalog identified the heterogeneity of escape across cell types and tissues.
Keywords: X chromosome; sex differences; single-cell omics.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
