

Gut microbiota dysbiosis is associated with altered tryptophan metabolism and dysregulated inflammatory response in COVID-19
- PMID: 39085233
- PMCID: PMC11291933
- DOI: 10.1038/s41522-024-00538-0
Gut microbiota dysbiosis is associated with altered tryptophan metabolism and dysregulated inflammatory response in COVID-19
Abstract
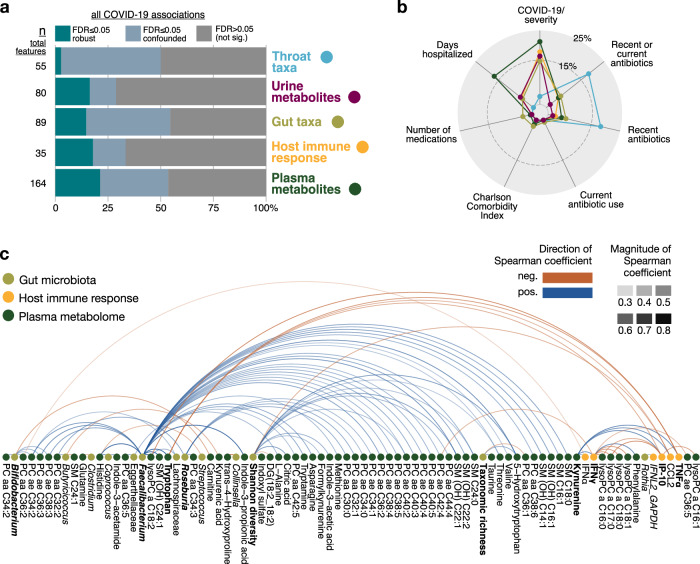
The clinical course of COVID-19 is variable and often unpredictable. To test the hypothesis that disease progression and inflammatory responses associate with alterations in the microbiome and metabolome, we analyzed metagenome, metabolome, cytokine, and transcriptome profiles of repeated samples from hospitalized COVID-19 patients and uninfected controls, and leveraged clinical information and post-hoc confounder analysis. Severe COVID-19 was associated with a depletion of beneficial intestinal microbes, whereas oropharyngeal microbiota disturbance was mainly linked to antibiotic use. COVID-19 severity was also associated with enhanced plasma concentrations of kynurenine and reduced levels of several other tryptophan metabolites, lysophosphatidylcholines, and secondary bile acids. Moreover, reduced concentrations of various tryptophan metabolites were associated with depletion of Faecalibacterium, and tryptophan decrease and kynurenine increase were linked to enhanced production of inflammatory cytokines. Collectively, our study identifies correlated microbiome and metabolome alterations as a potential contributor to inflammatory dysregulation in severe COVID-19.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
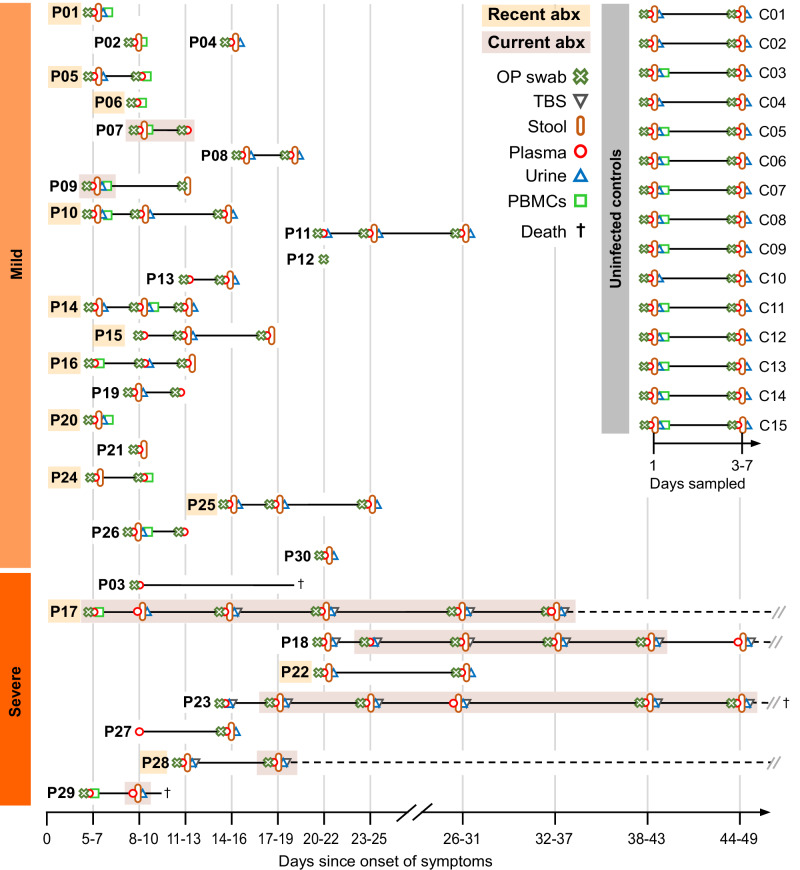
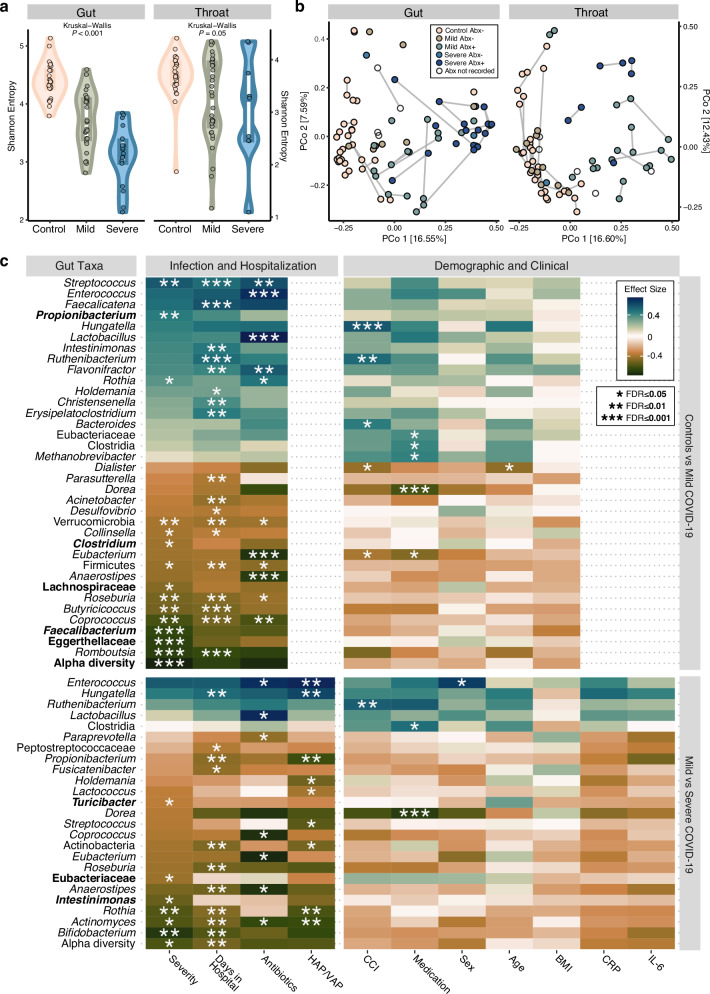
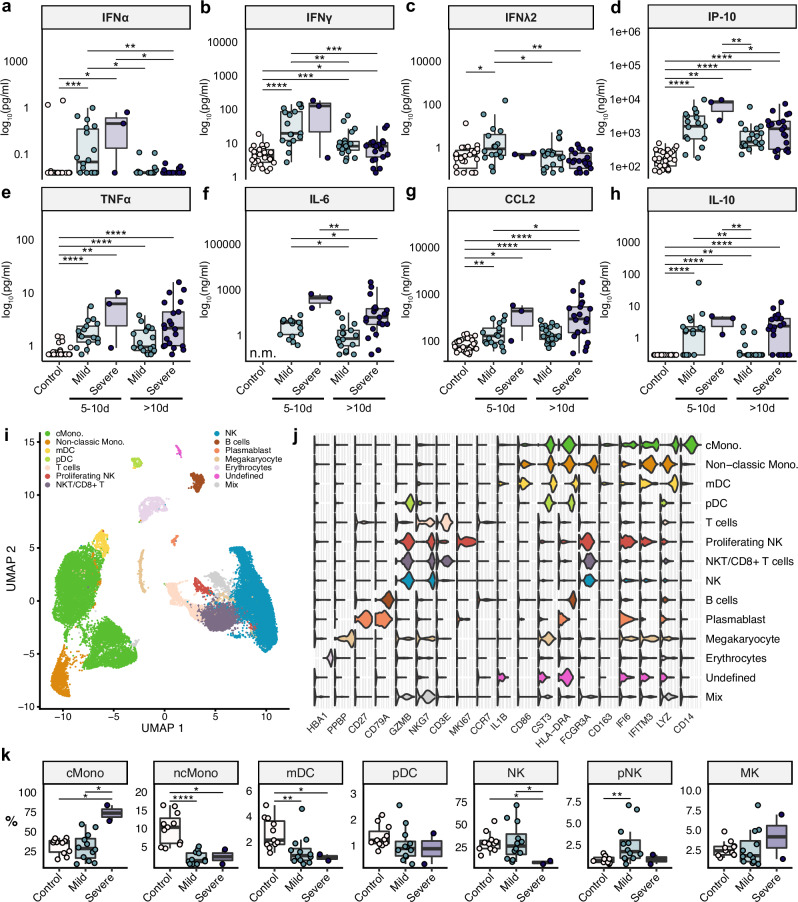
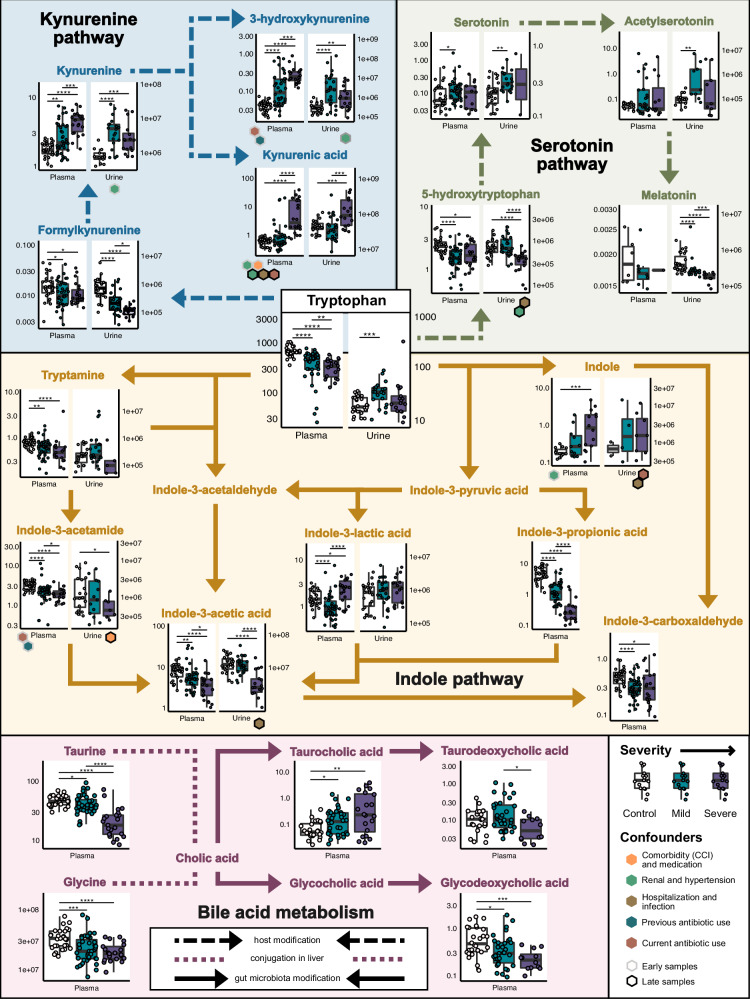
References
MeSH terms
Substances
Grants and funding
- SFB-TR84 A1/A5/Deutsche Forschungsgemeinschaft (German Research Foundation)
- 400667201/Deutsche Forschungsgemeinschaft (German Research Foundation)
- SFB-TR84/Deutsche Forschungsgemeinschaft (German Research Foundation)
- SFB-TR84/Deutsche Forschungsgemeinschaft (German Research Foundation)
- SFB1365, SFB1470, KFO339/Deutsche Forschungsgemeinschaft (German Research Foundation)
LinkOut - more resources
Full Text Sources
Medical
