

Biophysical cartography of the native and human-engineered antibody landscapes quantifies the plasticity of antibody developability
- PMID: 39085379
- PMCID: PMC11291509
- DOI: 10.1038/s42003-024-06561-3
Biophysical cartography of the native and human-engineered antibody landscapes quantifies the plasticity of antibody developability
Abstract
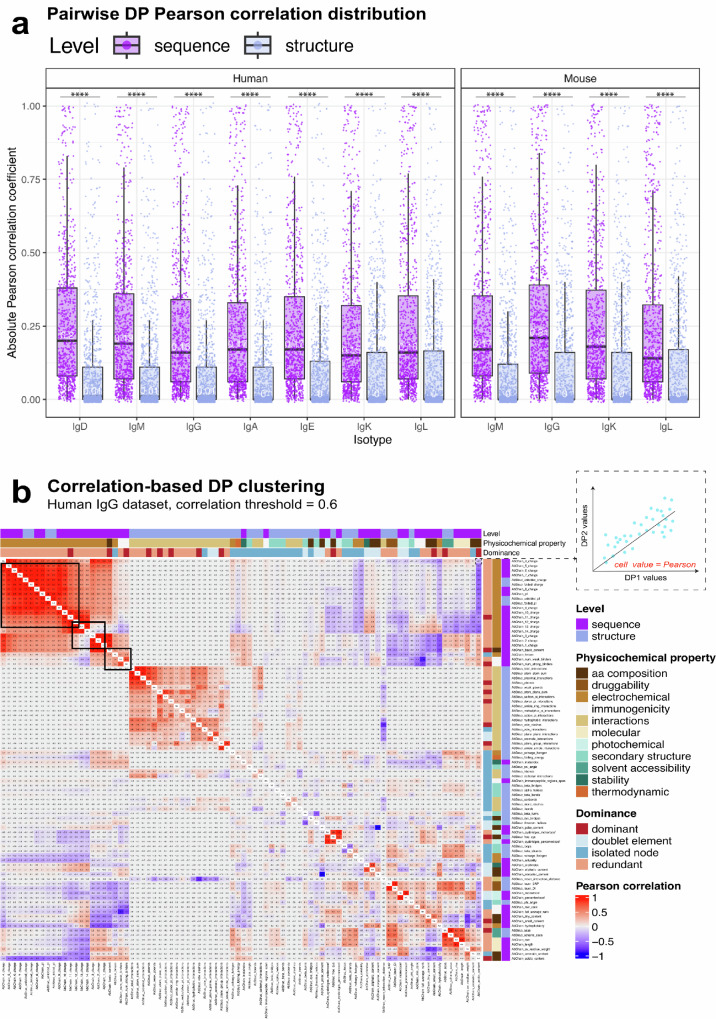
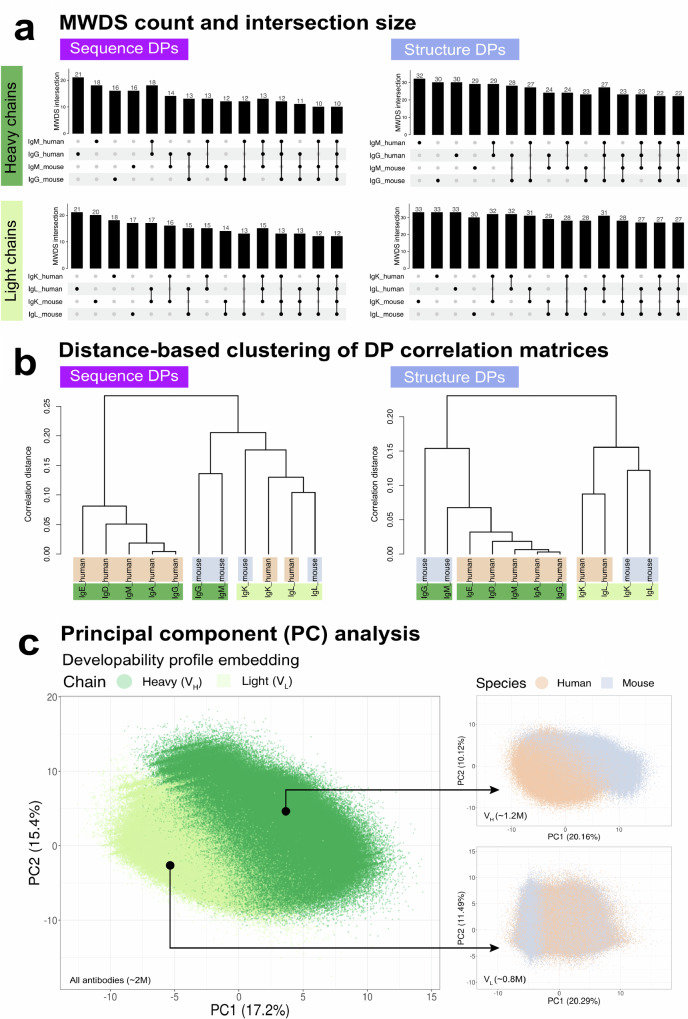
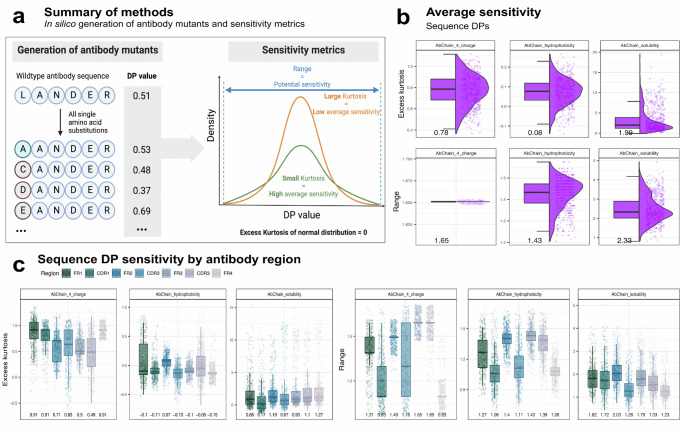
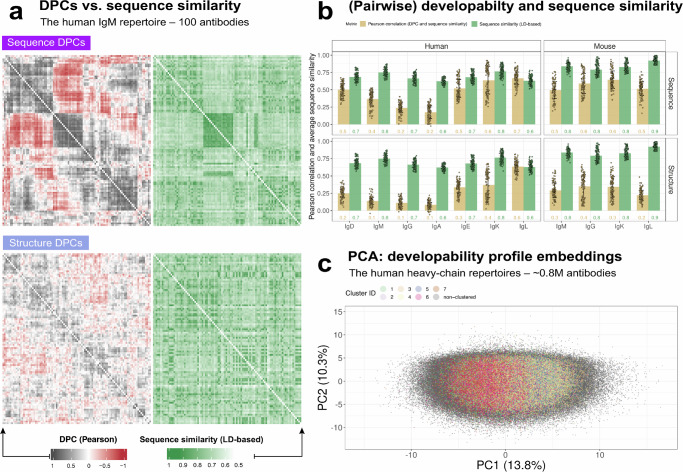
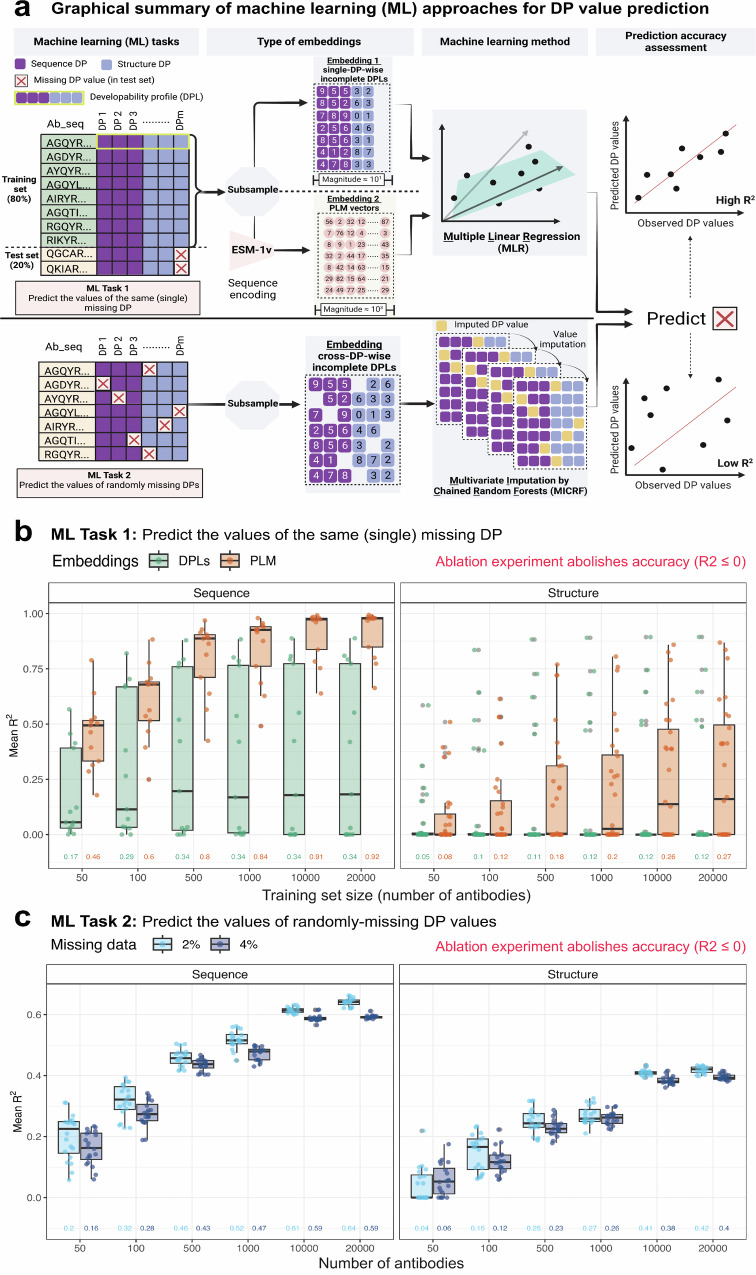
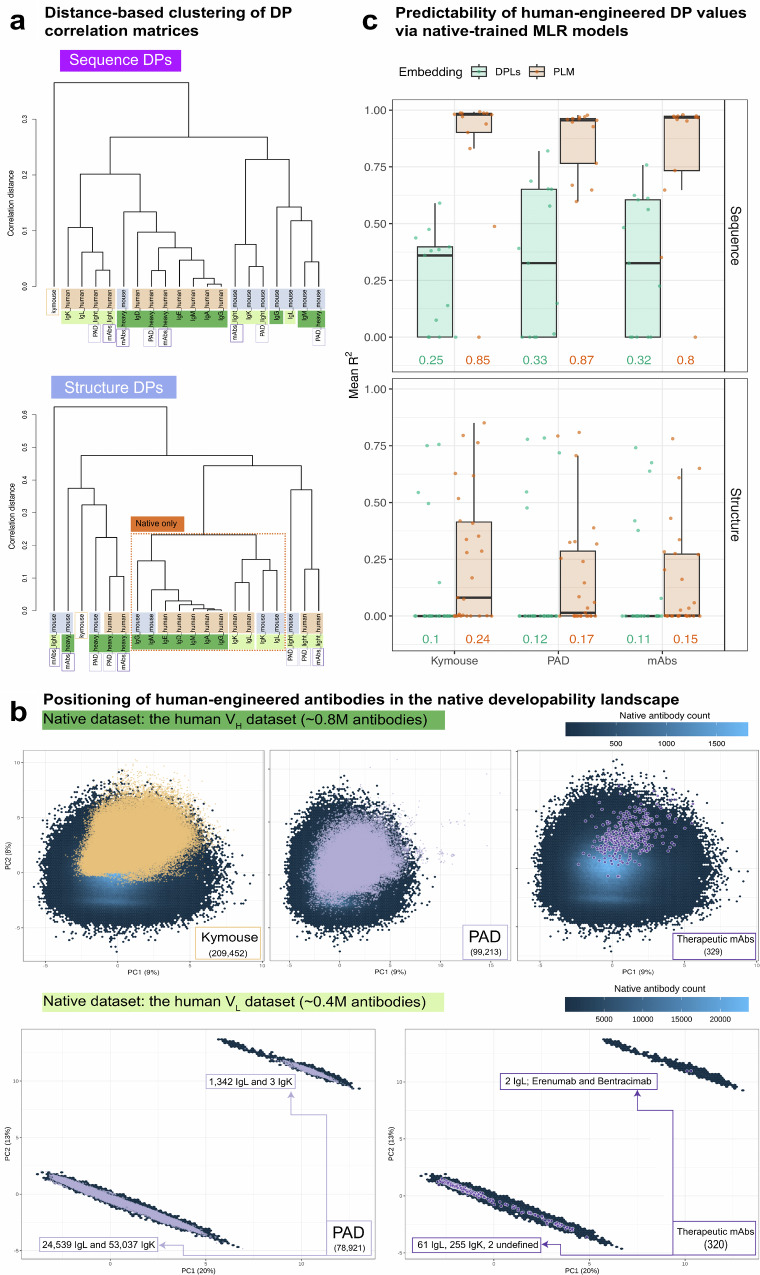
Designing effective monoclonal antibody (mAb) therapeutics faces a multi-parameter optimization challenge known as "developability", which reflects an antibody's ability to progress through development stages based on its physicochemical properties. While natural antibodies may provide valuable guidance for mAb selection, we lack a comprehensive understanding of natural developability parameter (DP) plasticity (redundancy, predictability, sensitivity) and how the DP landscapes of human-engineered and natural antibodies relate to one another. These gaps hinder fundamental developability profile cartography. To chart natural and engineered DP landscapes, we computed 40 sequence- and 46 structure-based DPs of over two million native and human-engineered single-chain antibody sequences. We find lower redundancy among structure-based compared to sequence-based DPs. Sequence DP sensitivity to single amino acid substitutions varied by antibody region and DP, and structure DP values varied across the conformational ensemble of antibody structures. We show that sequence DPs are more predictable than structure-based ones across different machine-learning tasks and embeddings, indicating a constrained sequence-based design space. Human-engineered antibodies localize within the developability and sequence landscapes of natural antibodies, suggesting that human-engineered antibodies explore mere subspaces of the natural one. Our work quantifies the plasticity of antibody developability, providing a fundamental resource for multi-parameter therapeutic mAb design.
© 2024. The Author(s).
Conflict of interest statement
The authors declare the following competing interests: V.G. declares advisory board positions in aiNET GmbH, Enpicom B.V, Absci, Omniscope, and Diagonal Therapeutics. V.G. is a consultant for Adaptyv Biosystems, Specifica Inc, Roche/Genentech, immunai, Proteinea and LabGenius. H.B. declares a scientific writing post in PipeBio ApS. K.K. is the founder of NaturalAntibody. M.P. and D.N-Z.G. are employed by Adaptyv Biosystems.
Figures
References
-
- Singh, S. et al. Monoclonal antibodies: a review. Curr. Clin. Pharmacol.13, 85–99 (2018). - PubMed
-
- Laustsen, A. H., Greiff, V., Karatt-Vellatt, A., Muyldermans, S. & Jenkins, T. P. Animal immunization, in vitro display technologies, and machine learning for antibody discovery. Trends Biotechnol. 39, 1263–1273 (2021). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
