

Polymersomes with splenic avidity target red pulp myeloid cells for cancer immunotherapy
- PMID: 39085390
- PMCID: PMC11567884
- DOI: 10.1038/s41565-024-01727-w
Polymersomes with splenic avidity target red pulp myeloid cells for cancer immunotherapy
Abstract
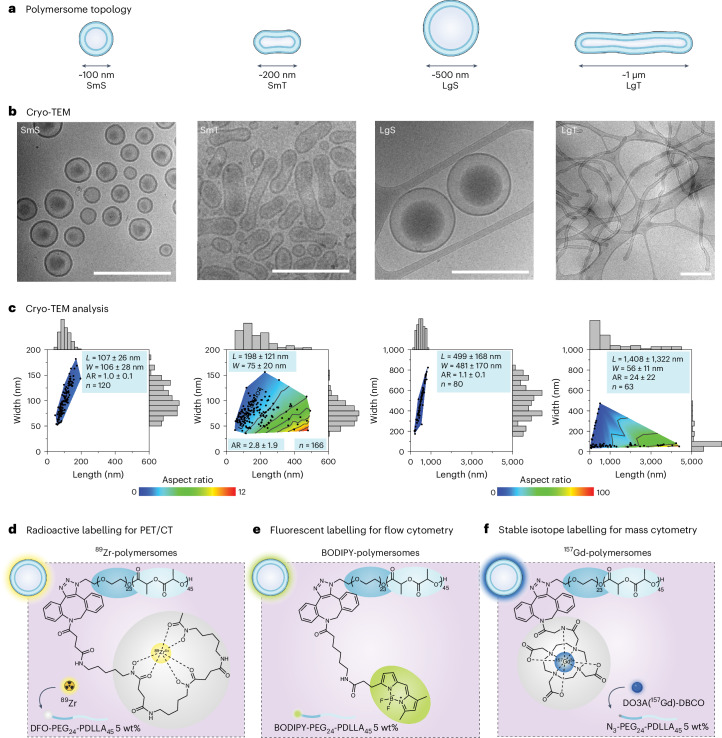
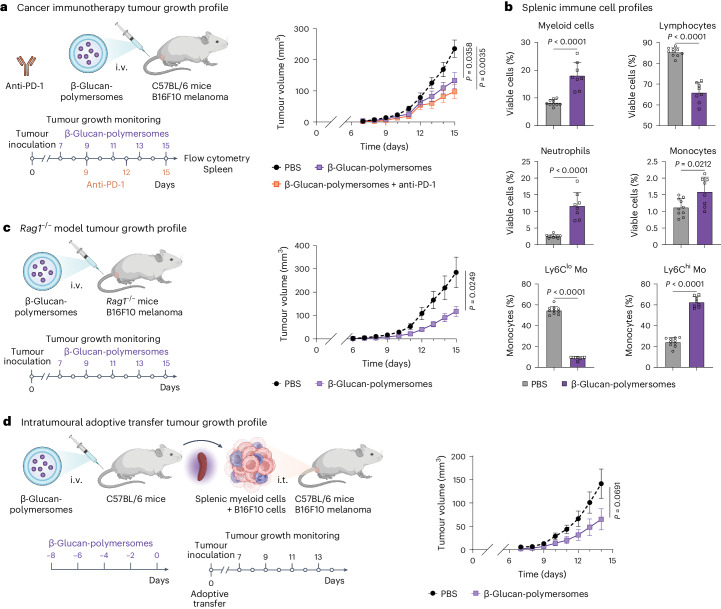
Regulating innate immunity is an emerging approach to improve cancer immunotherapy. Such regulation requires engaging myeloid cells by delivering immunomodulatory compounds to hematopoietic organs, including the spleen. Here we present a polymersome-based nanocarrier with splenic avidity and propensity for red pulp myeloid cell uptake. We characterized the in vivo behaviour of four chemically identical yet topologically different polymersomes by in vivo positron emission tomography imaging and innovative flow and mass cytometry techniques. Upon intravenous administration, relatively large and spherical polymersomes accumulated rapidly in the spleen and efficiently targeted myeloid cells in the splenic red pulp. When loaded with β-glucan, intravenously administered polymersomes significantly reduced tumour growth in a mouse melanoma model. We initiated our nanotherapeutic's clinical translation with a biodistribution study in non-human primates, which revealed that the platform's splenic avidity is preserved across species.
© 2024. The Author(s).
Conflict of interest statement
Figures



References
-
- Theobald, M. (ed). Current Immunotherapeutic Strategies in Cancer (Springer, 2020).
MeSH terms
Substances
Grants and funding
- Baroness Ariane de Rothschild Women Doctoral Program/Rothschild Caesarea Foundation
- P01 HL131478/HL/NHLBI NIH HHS/United States
- ERC Advanced Grant (#833247)/EC | Horizon 2020 Framework Programme (EU Framework Programme for Research and Innovation H2020)
- ERC, Smart Nanoparticles, 101089009/EC | Horizon 2020 Framework Programme (EU Framework Programme for Research and Innovation H2020)
- postdoctoral fellowship/Azrieli Foundation
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
