

Antifibrotics and mortality in idiopathic pulmonary fibrosis: external validity and avoidance of immortal time bias
- PMID: 39085869
- PMCID: PMC11293013
- DOI: 10.1186/s12931-024-02922-y
Antifibrotics and mortality in idiopathic pulmonary fibrosis: external validity and avoidance of immortal time bias
Abstract
Background and objective: Pooled analyses of previous randomized controlled trials reported that antifibrotics improved survival in patients with idiopathic pulmonary fibrosis (IPF), but the results were only based on short-term outcome data from selected patients who met strict criteria. Observational studies/meta-analyses also suggested that antifibrotics improve survival, but these studies failed to control for immortal time bias that considerably exaggerates drug effects. Therefore, whether antifibrotics truly improve long-term survival in patients with IPF in the real world remains undetermined and requires external validity.
Methods: We used data from the Japanese National Claims Database to estimate the intention-to-treat effect of antifibrotics on mortality. To address immortal time bias, we employed models treating antifibrotic initiation as a time-dependent covariate and target trial emulation (TTE), both incorporating new-user designs for antifibrotics and treating lung transplantation as a competing event.
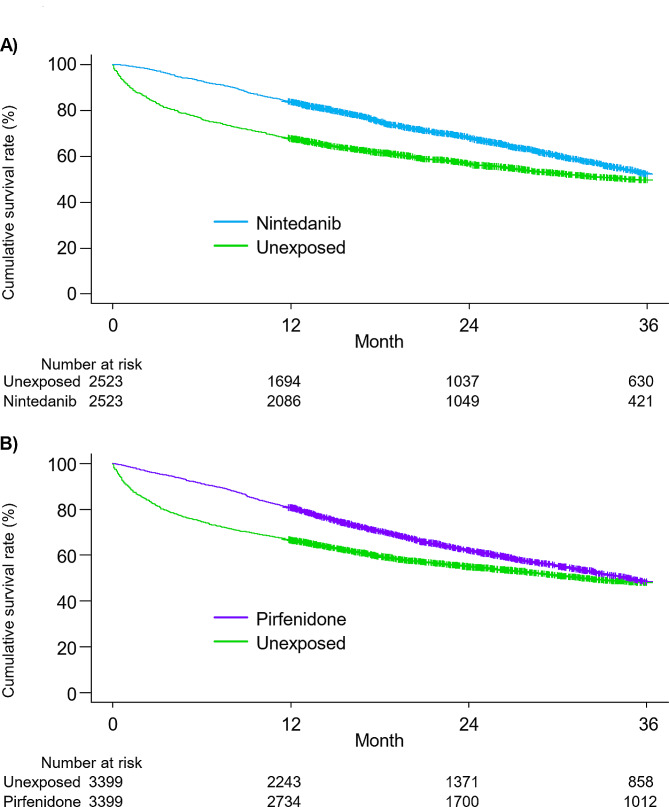
Results: Of 30,154 patients with IPF, 14,525 received antifibrotics. Multivariate Fine-Gray models with antifibrotic initiation as a time-dependent covariate revealed that compared with no treatment, nintedanib (adjusted hazard ratio [aHR], 0.85; 95% confidence interval [CI], 0.81-0.89) and pirfenidone (aHR, 0.89; 95% CI, 0.86-0.93) were associated with reduced mortality. The TTE model also replicated the associations of nintedanib (aHR, 0.69; 95% CI, 0.65-0.74) and pirfenidone (aHR, 0.81; 95% CI, 0.78-0.85) with reduced mortality. Subgroup analyses confirmed this association regardless of age, sex, and comorbidities, excluding certain subpopulations.
Conclusions: The results of this large-scale real-world analysis support the generalizability of the association between antifibrotics and improved survival in various IPF populations.
Keywords: Antifibrotics; Idiopathic pulmonary fibrosis; Mortality; Nintedanib; Pirfenidone.
© 2024. The Author(s).
Conflict of interest statement
Hozumi received honoraria for speaking engagements from Boehringer Ingelheim, which is not related to this manuscript. Suda received honoraria for speaking engagements from Boehringer Ingelheim and SHIONOGI & CO., LTD., but none of them are related to this manuscript. Miyashita, Nakatani, Inoue, Yasui, Suzuki, Karayama, Furuhashi, Enomoto, Fujisawa and Inui declare that no competing interests exist.
Figures


References
-
- Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, Kreuter M, Lynch DA, Maher TM, Martinez FJ, Molina-Molina M, Myers JL, Nicholson AG, Ryerson CJ, Strek ME, Troy LK, Wijsenbeek M, Mammen MJ, Hossain T, Bissell BD, Herman DD, Hon SM, Kheir F, Khor YH, Macrea M, Antoniou KM, Bouros D, Buendia-Roldan I, Caro F, Crestani B, Ho L, Morisset J, Olson AL, Podolanczuk A, Poletti V, Selman M, Ewing T, Jones S, Knight SL, Ghazipura M, Wilson KC. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2022;205:e18-e47. - PMC - PubMed
-
- Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, King TE Jr., Lancaster L, Sahn SA, Szwarcberg J, Valeyre D, du Bois RM. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet (London England). 2011;377:1760–9. 10.1016/S0140-6736(11)60405-4 - DOI - PubMed
-
- King TE Jr., Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, Lederer DJ, Nathan SD, Pereira CA, Sahn SA, Sussman R, Swigris JJ, Noble PW. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–92. 10.1056/NEJMoa1402582 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
