

Case report: Variability in clinical manifestations within a family with incontinentia pigmenti
- PMID: 39086952
- PMCID: PMC11288940
- DOI: 10.3389/fmed.2024.1402577
Case report: Variability in clinical manifestations within a family with incontinentia pigmenti
Abstract
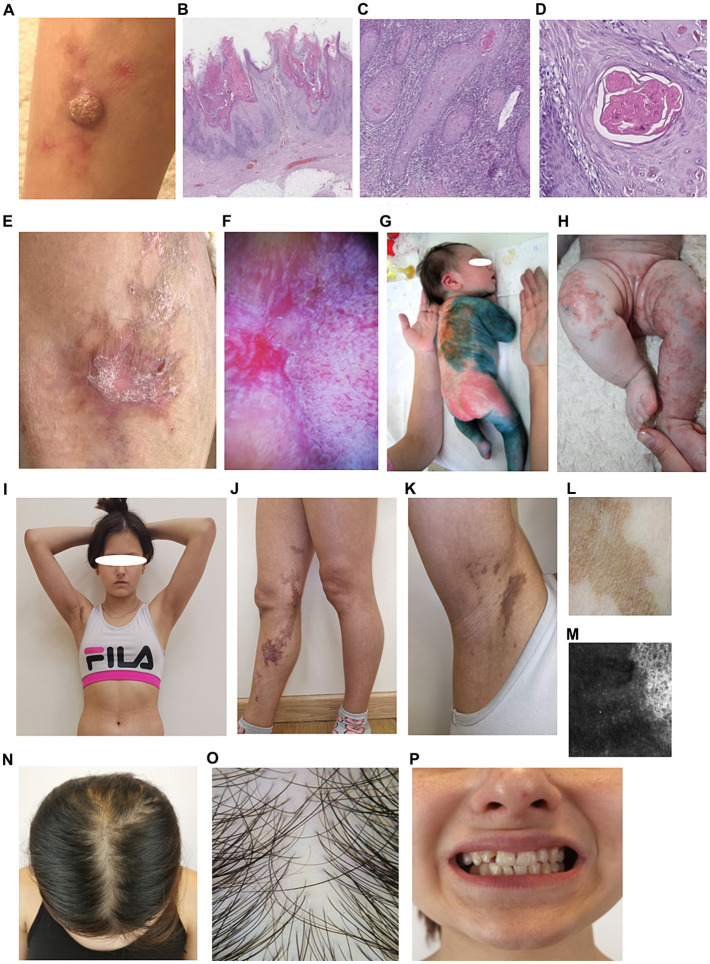
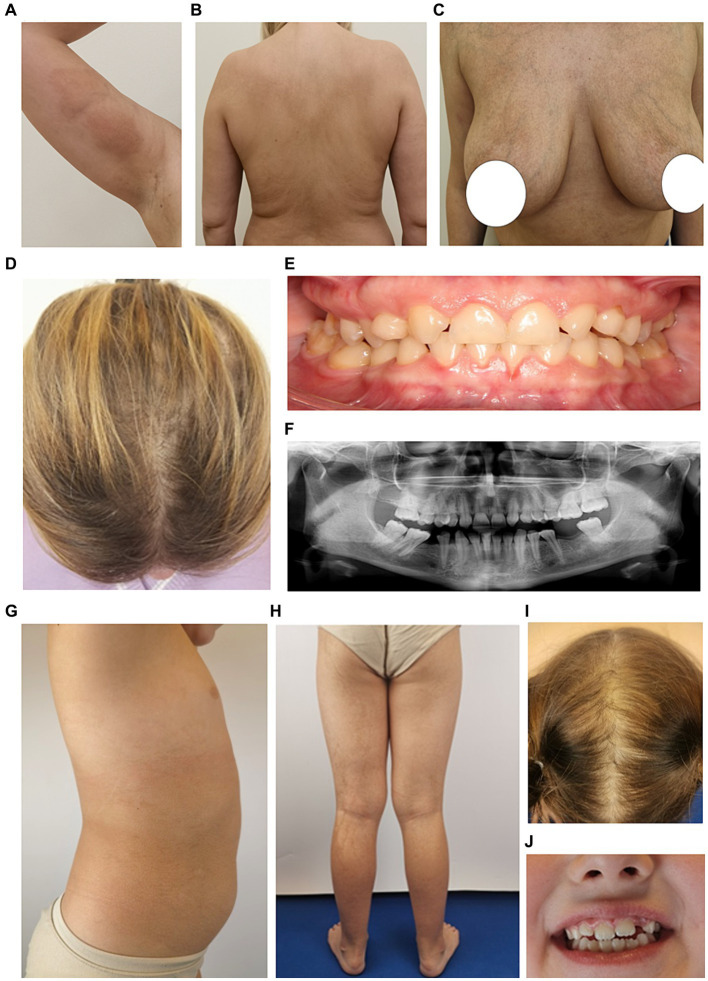
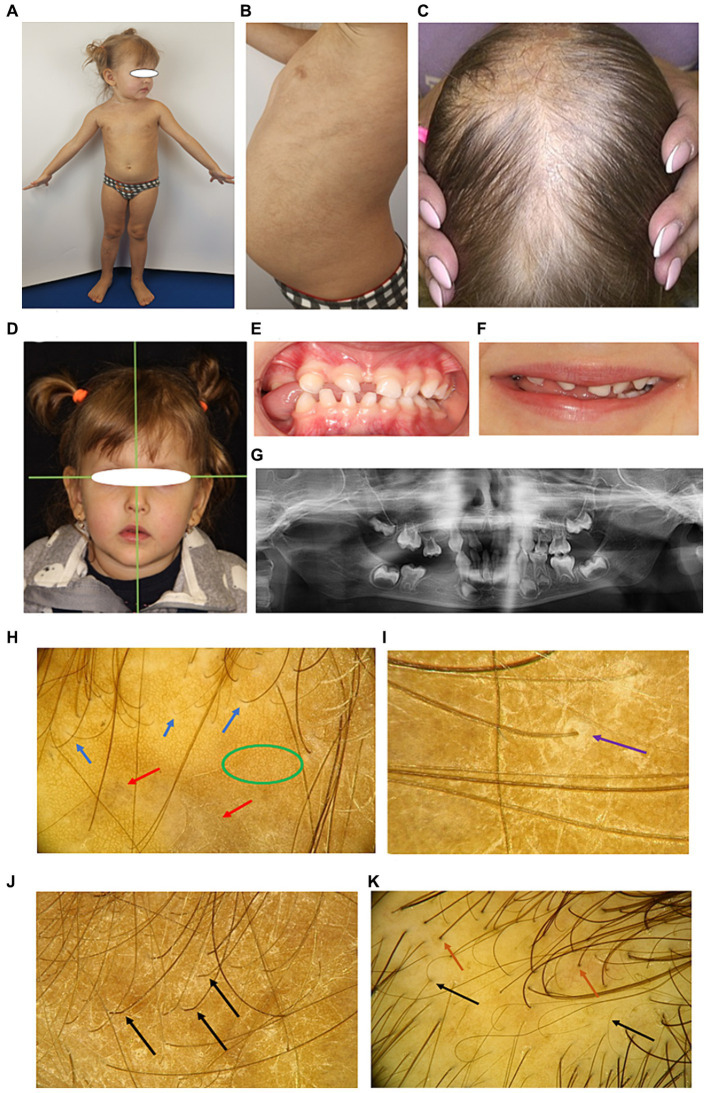
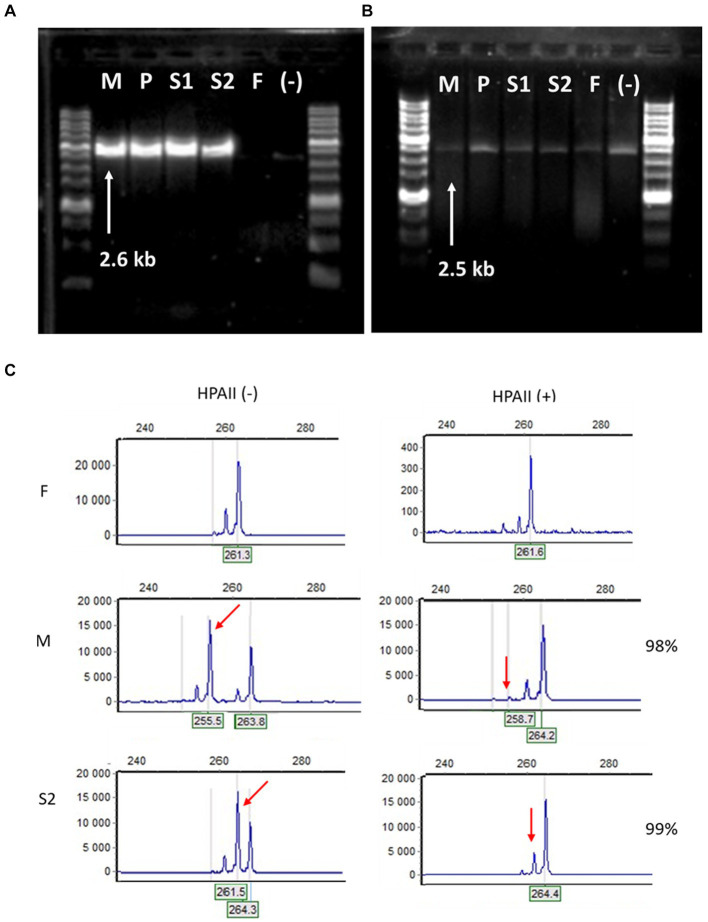
Diagnosing skin diseases in children can be a complex interdisciplinary problem. Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome, is a rare hereditary genodermatosis related to a mutation in the IKBKG gene. We present a family case of IP described from the perspective of various specialists, including dermatologists, oncologists, geneticists, dentists, and trichologists. The peculiarity of this case is the development of squamous cell carcinoma (SCC) on the shin of a 10-year-old female patient with IP. The patient had a positive family history: her mother and two sisters also displayed clinical manifestations of IP with involvement of skin, teeth and hair. The presence of exons 4-10 deletion in the IKBKG gene in all affected females was confirmed by detailed genetic evaluation using long-range PCR, and also high degree of X-chromosome inactivation skewing was demonstrated. The family underwent a comprehensive examination and was followed up for 2 years with successful symptomatic treatment of dermatologic manifestations. Recommendations were also made regarding dental and hair problems. By the end of the follow-up period, patients had stabilized, with the exception of a 36-year-old mother who developed generalized morphea. The study demonstrates the varying expressiveness of clinical symptoms among family members and emphasizes the importance of timely diagnosis for effective management of patients with IP.
Keywords: IKBKG/NEMO deletion; X-chromosome inactivation; dental abnormalities; family case report; hair; incontinentia pigmenti; squamous cell carcinoma.
Copyright © 2024 Belysheva, Nasedkina, Kletskaya, Volchek, Barinova, Semenova, Gadzhigoroeva, Zelenova, Valiev, Sharapova, Michenko, Allenova and Ponomareva.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Incontinentia Pigmenti.Actas Dermosifiliogr (Engl Ed). 2019 May;110(4):273-278. doi: 10.1016/j.ad.2018.10.004. Epub 2019 Jan 17. Actas Dermosifiliogr (Engl Ed). 2019. PMID: 30660327 Review. English, Spanish.
-
Incontinentia Pigmenti (Bloch-Sulzberger Syndrome).2023 Aug 25. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2023 Aug 25. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 35201722 Free Books & Documents.
-
Importance of extracutaneous organ involvement in determining the clinical severity and prognosis of incontinentia pigmenti caused by mutations in the IKBKG gene.Exp Dermatol. 2021 May;30(5):676-683. doi: 10.1111/exd.14313. Epub 2021 Mar 10. Exp Dermatol. 2021. PMID: 33655605
-
[Dental anomalies associated with incontinentia pigmenti or Bloch-Sulzberger syndrome].Rev Belge Med Dent (1984). 2004;59(2):94-9. Rev Belge Med Dent (1984). 2004. PMID: 15690773 French.
-
Incontinentia pigmenti (Bloch-Sulzberger syndrome): a systemic disorder.Cutis. 2007 May;79(5):355-62. Cutis. 2007. PMID: 17569396 Review.
References
-
- Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, et al. . Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The international Incontinentia Pigmenti (IP) consortium. Nature. (2000) 405:466–72. doi: 10.1038/35013114, PMID: - DOI - PubMed
-
- Fusco F, Pescatore A, Conte MI, Mirabelli P, Paciolla M, Esposito E, et al. . EDA-ID and IP, two faces of the same coin: how the same IKBKG/NEMO mutation affecting the NF-κB pathway can cause immunodeficiency and/or inflammation. Int Rev Immunol. (2015) 34:445–59. doi: 10.3109/08830185.2015.1055331 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous