

The parkin V380L variant is a genetic modifier of Machado-Joseph disease with impact on mitophagy
- PMID: 39088078
- PMCID: PMC11294389
- DOI: 10.1007/s00401-024-02762-6
The parkin V380L variant is a genetic modifier of Machado-Joseph disease with impact on mitophagy
Abstract
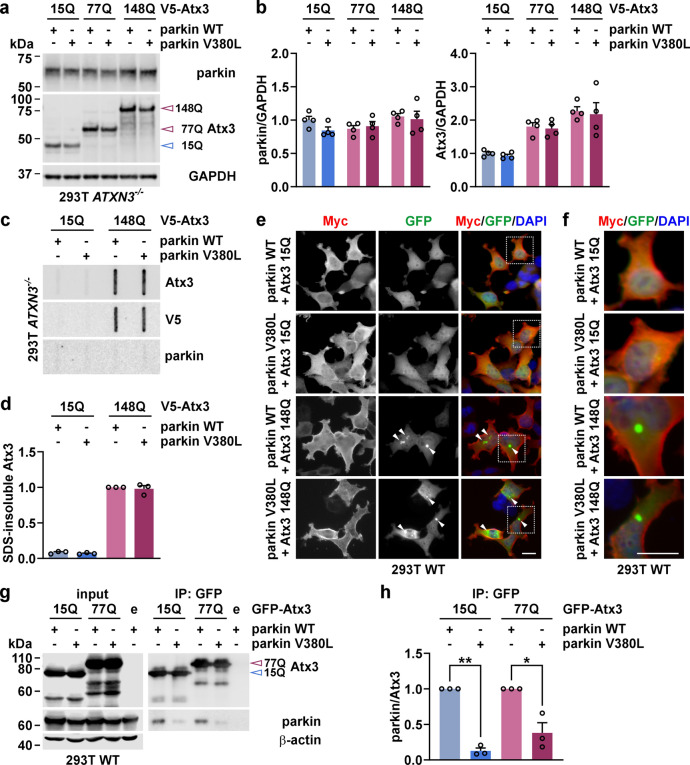
Machado-Joseph disease (MJD) is an autosomal dominant neurodegenerative spinocerebellar ataxia caused by a polyglutamine-coding CAG repeat expansion in the ATXN3 gene. While the CAG length correlates negatively with the age at onset, it accounts for approximately 50% of its variability only. Despite larger efforts in identifying contributing genetic factors, candidate genes with a robust and plausible impact on the molecular pathogenesis of MJD are scarce. Therefore, we analysed missense single nucleotide polymorphism variants in the PRKN gene encoding the Parkinson's disease-associated E3 ubiquitin ligase parkin, which is a well-described interaction partner of the MJD protein ataxin-3, a deubiquitinase. By performing a correlation analysis in the to-date largest MJD cohort of more than 900 individuals, we identified the V380L variant as a relevant factor, decreasing the age at onset by 3 years in homozygous carriers. Functional analysis in an MJD cell model demonstrated that parkin V380L did not modulate soluble or aggregate levels of ataxin-3 but reduced the interaction of the two proteins. Moreover, the presence of parkin V380L interfered with the execution of mitophagy-the autophagic removal of surplus or damaged mitochondria-thereby compromising cell viability. In summary, we identified the V380L variant in parkin as a genetic modifier of MJD, with negative repercussions on its molecular pathogenesis and disease age at onset.
Keywords: PRKN; Aggregation; Polyglutamine disease; SCA3; SNP; Spinocerebellar ataxia type 3.
© 2024. The Author(s).
Conflict of interest statement
The authors have no relevant financial or non-financial interests to disclose.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
- DFG/Deutsche Forschungsgemeinschaft
- WE 6585/1-1/Deutsche Forschungsgemeinschaft
- CNPq/Ciência sem Fronteiras
- Process no. 229957/2013-7/Ciência sem Fronteiras
- ERN-RND/European Reference Network for Rare Neurological Diseases
- Project ID no. 739510/European Reference Network for Rare Neurological Diseases
- BMBF/Bundesministerium für Bildung und Forschung
- E-Rare project SCA-CYP/Bundesministerium für Bildung und Forschung
- grant no. 01GM1803; EuSAge project/Bundesministerium für Bildung und Forschung
- grant no. 01DN18020/Bundesministerium für Bildung und Forschung
- EUROSCA/European commission, FP6
- LSHM-CT-2004-503304/European commission, FP6
LinkOut - more resources
Full Text Sources
