

Data acquisition approaches for single cell proteomics
- PMID: 39088833
- PMCID: PMC11735665
- DOI: 10.1002/pmic.202400022
Data acquisition approaches for single cell proteomics
Abstract
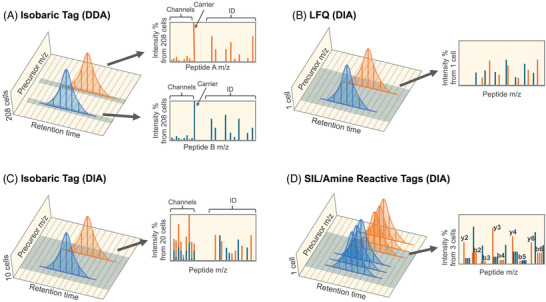
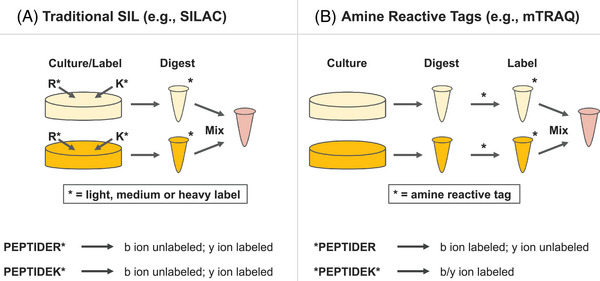
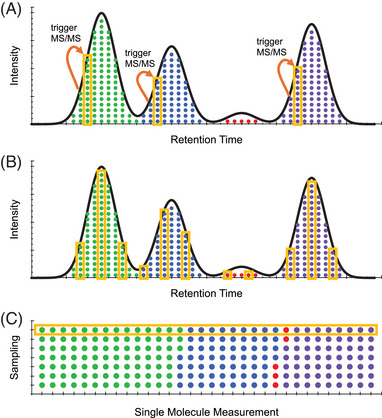
Single-cell proteomics (SCP) aims to characterize the proteome of individual cells, providing insights into complex biological systems. It reveals subtle differences in distinct cellular populations that bulk proteome analysis may overlook, which is essential for understanding disease mechanisms and developing targeted therapies. Mass spectrometry (MS) methods in SCP allow the identification and quantification of thousands of proteins from individual cells. Two major challenges in SCP are the limited material in single-cell samples necessitating highly sensitive analytical techniques and the efficient processing of samples, as each biological sample requires thousands of single cell measurements. This review discusses MS advancements to mitigate these challenges using data-dependent acquisition (DDA) and data-independent acquisition (DIA). Additionally, we examine the use of short liquid chromatography gradients and sample multiplexing methods that increase the sample throughput and scalability of SCP experiments. We believe these methods will pave the way for improving our understanding of cellular heterogeneity and its implications for systems biology.
Keywords: data dependent acquisition; data independent acquisition; mass spectrometry; multiplex; proteomics; single cell.
© 2024 The Author(s). PROTEOMICS published by Wiley‐VCH GmbH.
Conflict of interest statement
Brian C. Searle is a founder and shareholder in Proteome Software, which operates in the field of proteomics.
Figures
References
-
- Regev, A. , Teichmann, S. A. , Lander, E. S. , Amit, I. , Benoist, C. , Birney, E. , Bodenmiller, B. , Campbell, P. , Carninci, P. , Clatworthy, M. , Clevers, H. , Deplancke, B. , Dunham, I. , Eberwine, J. , Eils, R. , Enard, W. , Farmer, A. , Fugger, L. , Göttgens, B. , … The Human Cell Atlas . (2017). The human cell atlas. eLife, 6. 10.7554/eLife.27041 - DOI - PMC - PubMed
-
- Shlush, L. I. , Mitchell, A. , Heisler, L. , Abelson, S. , Ng, S. W. K. , Trotman‐Grant, A. , Medeiros, J. J. F. , Rao‐Bhatia, A. , Jaciw‐Zurakowsky, I. , Marke, R. , Mcleod, J. L. , Doedens, M. , Bader, G. , Voisin, V. , Xu, C. , Mcpherson, J. D. , Hudson, T. J. , Wang, J. C. Y. , Minden, M. D. , … Dick, J. E. (2017). Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature, 547(7661), 104–108. - PubMed
-
- Nam, A. S. , Kim, K. T. , Chaligne, R. , Izzo, F. , Ang, C. , Taylor, J. , Myers, R. M. , Abu‐Zeinah, G. , Brand, R. , Omans, N. D. , Alonso, A. , Sheridan, C. , Mariani, M. , Dai, X. , Harrington, E. , Pastore, A. , Cubillos‐Ruiz, J. R. , Tam, W. , Hoffman, R. , … Landau, D. A. (2019). Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature, 571(7765), 355–360. - PMC - PubMed
-
- Kreso, A. , & Dick, J. E. (2014). Evolution of the cancer stem cell model. Cell Stem Cell, 14(3), 275–291. - PubMed
-
- Ramsköld, D. , Luo, S. , Wang, Y.‐C. , Li, R. , Deng, Q. , Faridani, O. R. , Daniels, G. A. , Khrebtukova, I. , Loring, J. F. , Laurent, L. C. , Schroth, G. P. , & Sandberg, R. (2012). Full‐length mRNA‐Seq from single‐cell levels of RNA and individual circulating tumor cells. Nature Biotechnology, 30(8), 777–782. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
