

Molecular mechanism of distinct chemokine engagement and functional divergence of the human Duffy antigen receptor
- PMID: 39089252
- PMCID: PMC11349380
- DOI: 10.1016/j.cell.2024.07.005
Molecular mechanism of distinct chemokine engagement and functional divergence of the human Duffy antigen receptor
Abstract
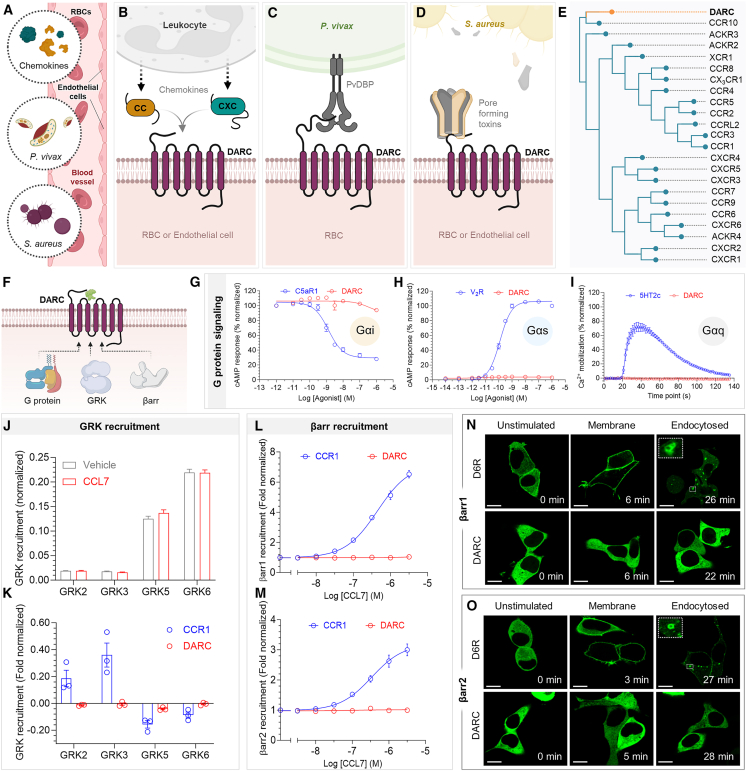
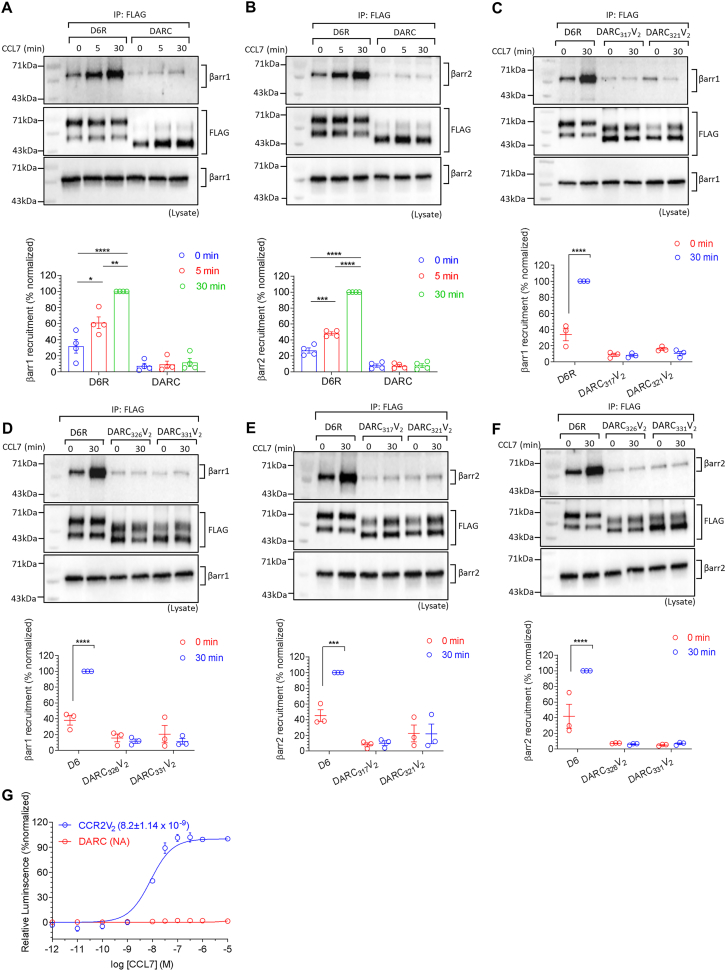
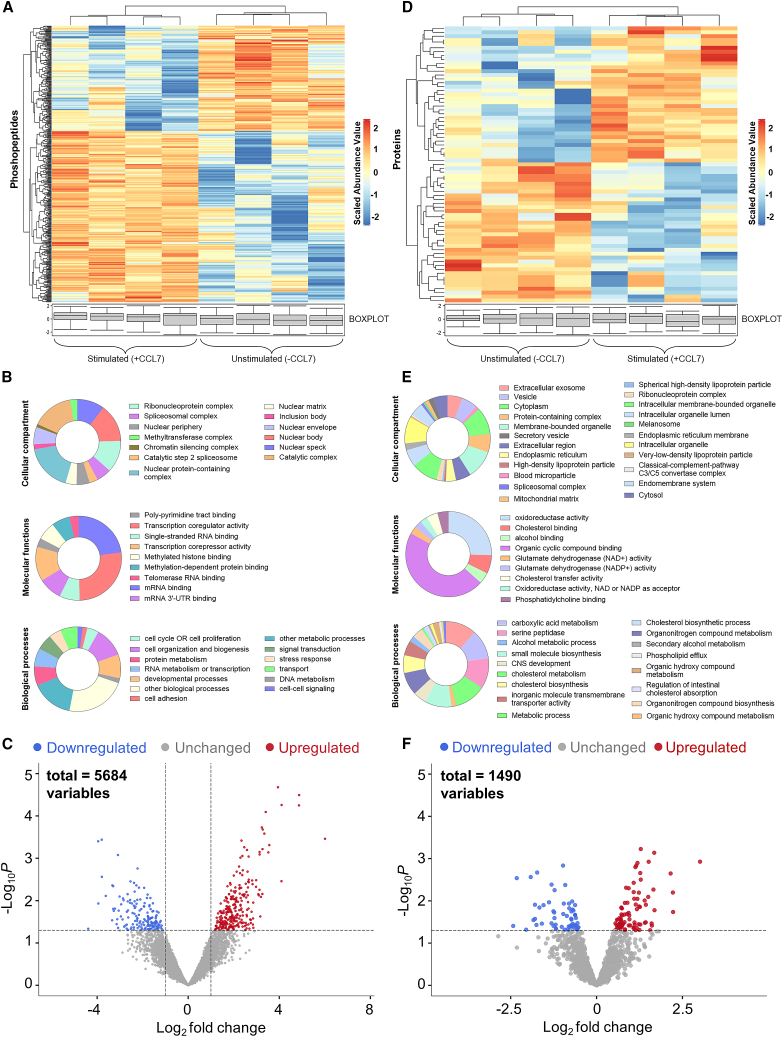
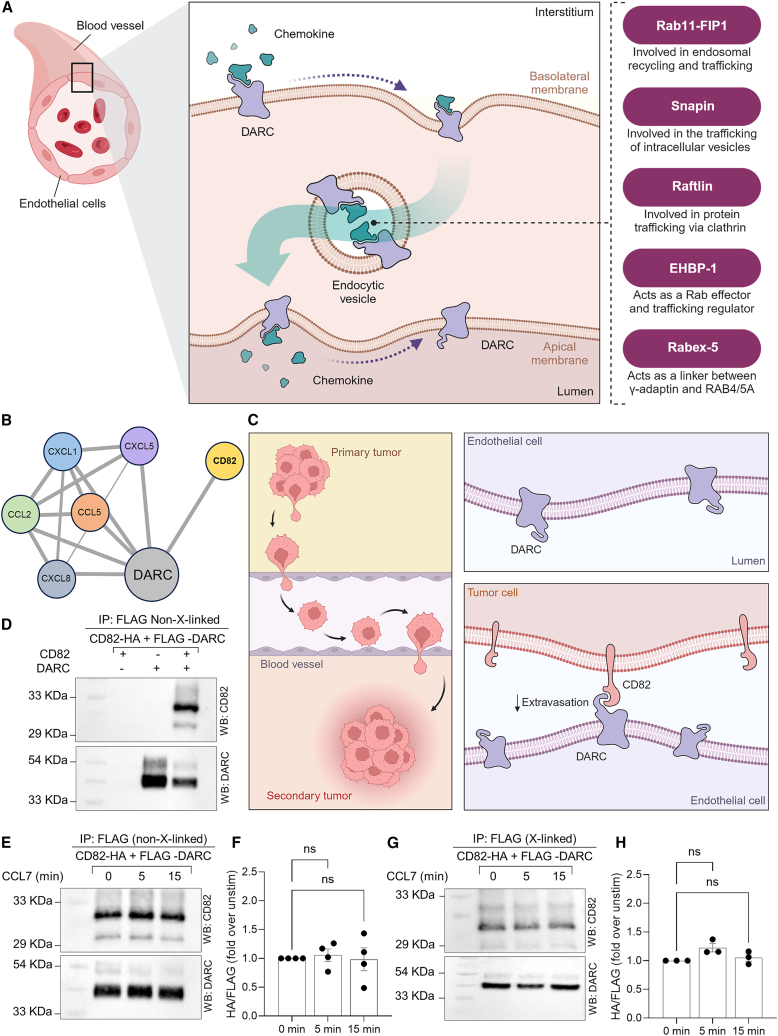
The Duffy antigen receptor is a seven-transmembrane (7TM) protein expressed primarily at the surface of red blood cells and displays strikingly promiscuous binding to multiple inflammatory and homeostatic chemokines. It serves as the basis of the Duffy blood group system in humans and also acts as the primary attachment site for malarial parasite Plasmodium vivax and pore-forming toxins secreted by Staphylococcus aureus. Here, we comprehensively profile transducer coupling of this receptor, discover potential non-canonical signaling pathways, and determine the cryoelectron microscopy (cryo-EM) structure in complex with the chemokine CCL7. The structure reveals a distinct binding mode of chemokines, as reflected by relatively superficial binding and a partially formed orthosteric binding pocket. We also observe a dramatic shortening of TM5 and 6 on the intracellular side, which precludes the formation of the docking site for canonical signal transducers, thereby providing a possible explanation for the distinct pharmacological and functional phenotype of this receptor.
Keywords: ACKR1; Duffy antigen receptor; G protein-coupled receptors; atypical chemokine receptors; cellular signaling; chemokine receptors; cryogenic-electron microscopy.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures






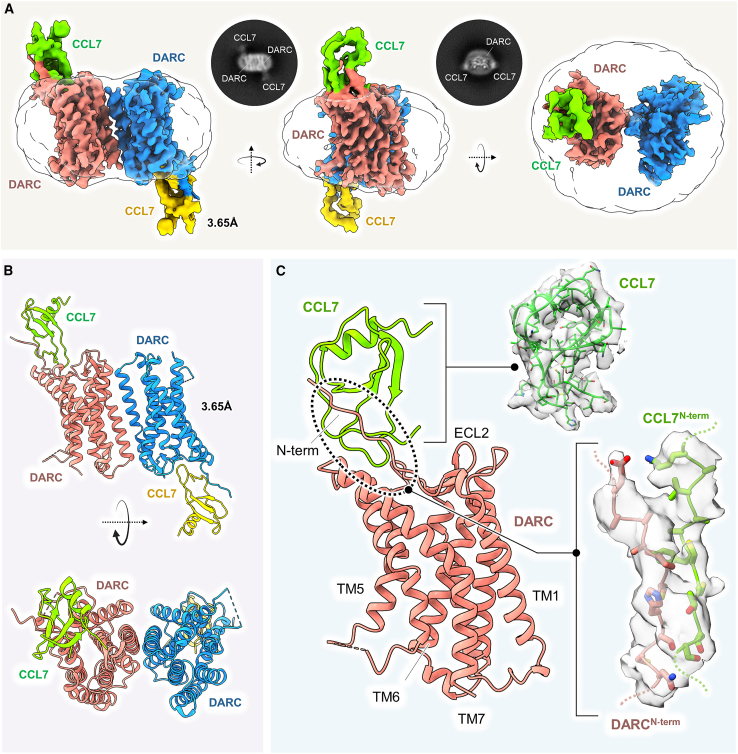

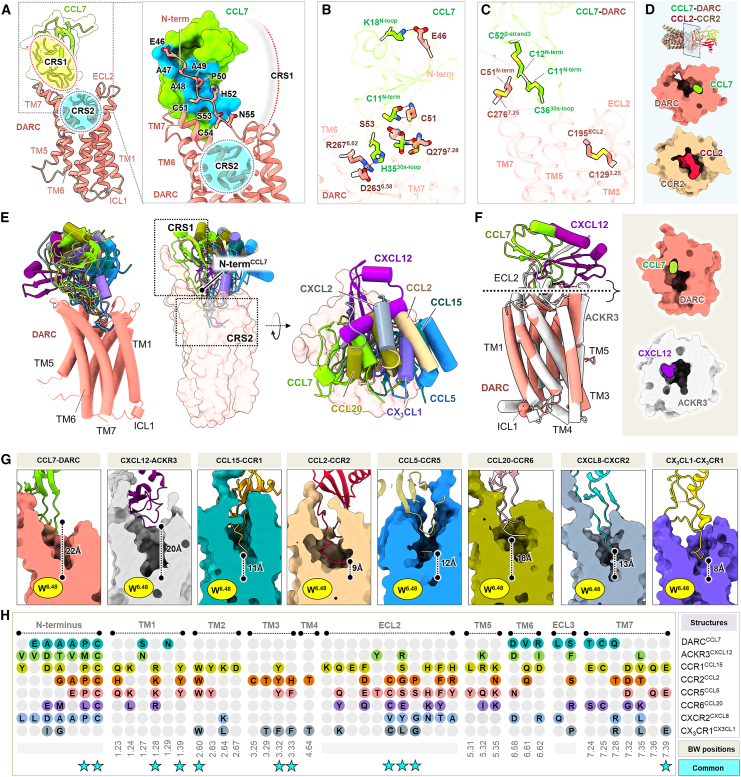



References
-
- Chaudhuri A., Polyakova J., Zbrzezna V., Williams K., Gulati S., Pogo A.O. Cloning of Glycoprotein D Cdna, Which Encodes the Major Subunit of the Duffy Blood-Group System and the Receptor for the Plasmodium-Vivax Malaria Parasite. Proc. Natl. Acad. Sci. USA. 1993;90:10793–10797. doi: 10.1073/pnas.90.22.10793. - DOI - PMC - PubMed
-
- Hadley T.J., Lu Z.H., Wasniowska K., Martin A.W., Peiper S.C., Hesselgesser J., Horuk R. Postcapillary venule endothelial cells in kidney express a multispecific chemokine receptor that is structurally and functionally identical to the erythroid isoform, which is the Duffy blood group antigen. J. Clin. Invest. 1994;94:985–991. doi: 10.1172/JCI117465. - DOI - PMC - PubMed
-
- Peiper S.C., Wang Z.X., Neote K., Martin A.W., Showell H.J., Conklyn M.J., Ogborne K., Hadley T.J., Lu Z.H., Hesselgesser J., et al. The Duffy antigen/receptor for chemokines (DARC) is expressed in endothelial cells of Duffy negative individuals who lack the erythrocyte receptor. J. Exp. Med. 1995;181:1311–1317. doi: 10.1084/jem.181.4.1311. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
