

Oxidative cell death in cancer: mechanisms and therapeutic opportunities
- PMID: 39090114
- PMCID: PMC11294602
- DOI: 10.1038/s41419-024-06939-5
Oxidative cell death in cancer: mechanisms and therapeutic opportunities
Abstract
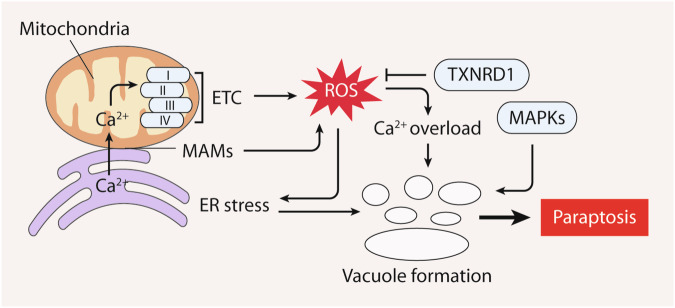
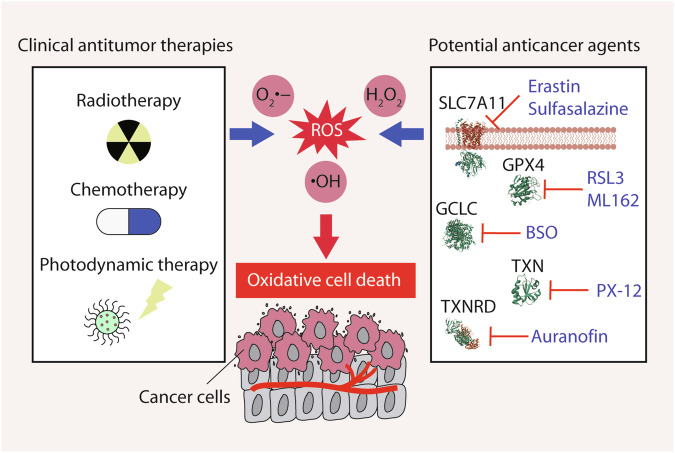
Reactive oxygen species (ROS) are highly reactive oxygen-containing molecules generated as natural byproducts during cellular processes, including metabolism. Under normal conditions, ROS play crucial roles in diverse cellular functions, including cell signaling and immune responses. However, a disturbance in the balance between ROS production and cellular antioxidant defenses can lead to an excessive ROS buildup, causing oxidative stress. This stress damages essential cellular components, including lipids, proteins, and DNA, potentially culminating in oxidative cell death. This form of cell death can take various forms, such as ferroptosis, apoptosis, necroptosis, pyroptosis, paraptosis, parthanatos, and oxeiptosis, each displaying distinct genetic, biochemical, and signaling characteristics. The investigation of oxidative cell death holds promise for the development of pharmacological agents that are used to prevent tumorigenesis or treat established cancer. Specifically, targeting key antioxidant proteins, such as SLC7A11, GCLC, GPX4, TXN, and TXNRD, represents an emerging approach for inducing oxidative cell death in cancer cells. This review provides a comprehensive summary of recent progress, opportunities, and challenges in targeting oxidative cell death for cancer therapy.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures






References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
