

This is a preprint.
The evolutionary fate of Neanderthal DNA in 30,780 admixed genomes with recent African-like ancestry
- PMID: 39091830
- PMCID: PMC11291122
- DOI: 10.1101/2024.07.25.605203
The evolutionary fate of Neanderthal DNA in 30,780 admixed genomes with recent African-like ancestry
Abstract
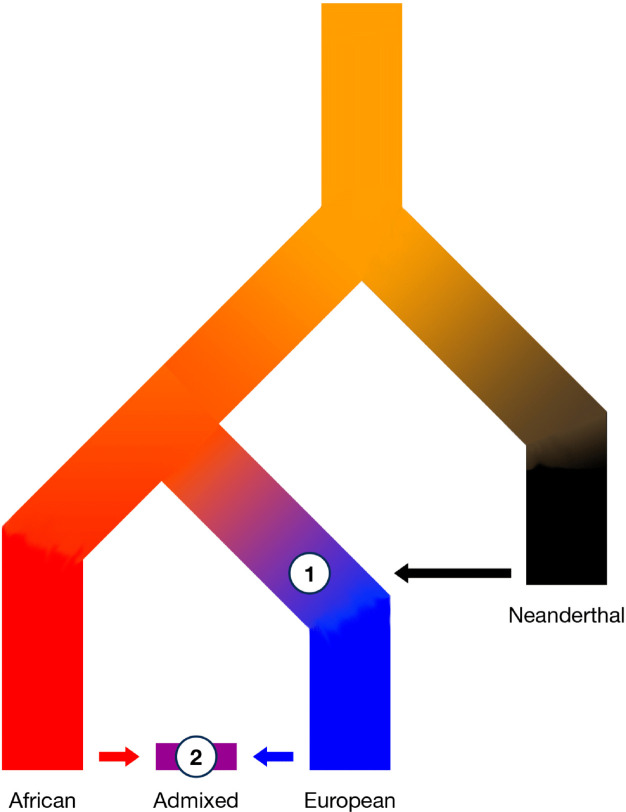
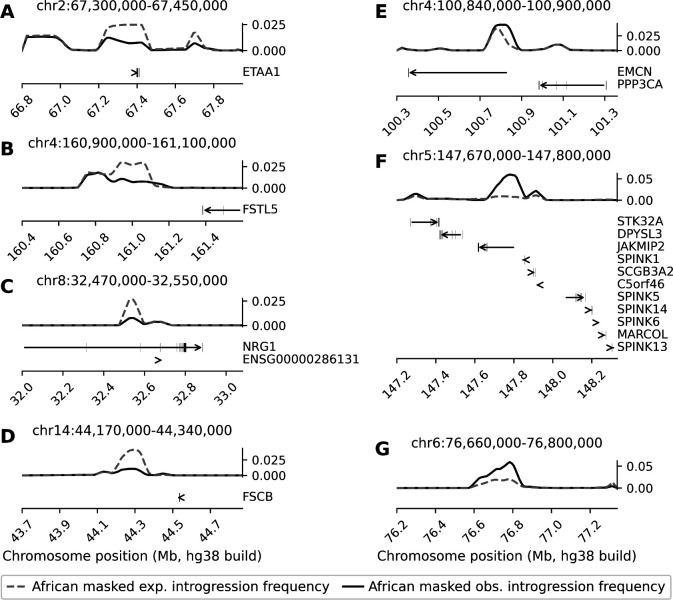
Following introgression, Neanderthal DNA was initially purged from non-African genomes, but the evolutionary fate of remaining introgressed DNA has not been explored yet. To fill this gap, we analyzed 30,780 admixed genomes with African-like ancestry from the All of Us research program, in which Neanderthal alleles encountered novel genetic backgrounds during the last 15 generations. Observed amounts of Neanderthal DNA approximately match expectations based on ancestry proportions, suggesting neutral evolution. Nevertheless, we identified genomic regions that have significantly less or more Neanderthal ancestry than expected and are associated with spermatogenesis, innate immunity, and other biological processes. We also identified three novel introgression desert-like regions in recently admixed genomes, whose genetic features are compatible with hybrid incompatibilities and intrinsic negative selection. Overall, we find that much of the remaining Neanderthal DNA in human genomes is not under strong selection, and complex evolutionary dynamics have shaped introgression landscapes in our species.
Keywords: Admixture; Hybrid incompatibilities; Introgression; Natural Selection; Neanderthal.
Conflict of interest statement
7Declaration of interests The authors declare no competing interests.
Figures




References
-
- Agresti A, Coull BA (1998) Approximate is Better than “Exact” for Interval Estimation of Binomial Proportions. The American Statistician 52(2):119–126. 10.1080/00031305.1998.10480550 - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous