

The E3 ubiquitin ligase Herc1 modulates the response to nucleoside analogs in acute myeloid leukemia
- PMID: 39093953
- PMCID: PMC11497402
- DOI: 10.1182/bloodadvances.2023011540
The E3 ubiquitin ligase Herc1 modulates the response to nucleoside analogs in acute myeloid leukemia
Abstract
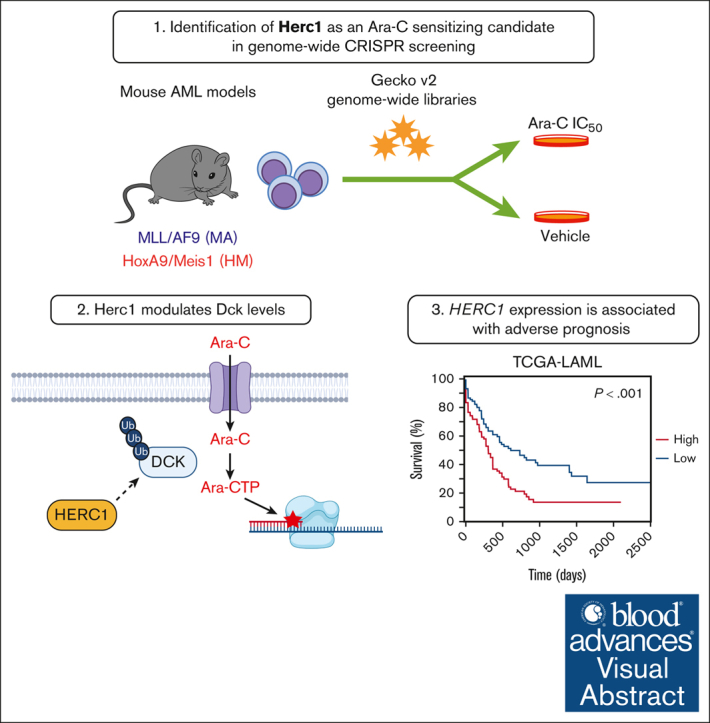
For several decades, induction therapy with nucleoside analogs, in particular cytarabine (Ara-C) and, to a lesser extent, fludarabine, has been the standard of care for patients diagnosed with acute myeloid leukemia (AML). However, the antitumor efficacy of nucleoside analogs is often limited by intrinsic and acquired drug resistance, thereby leading to poor therapeutic response and suboptimal clinical outcomes. In this study, we used genome-wide CRISPR-based pharmacogenomic screening to map the genetic factors that modulate the response to nucleoside analogs in AML and identified the E3 ubiquitin ligase, Herc1, as a key modulator of Ara-C response in mouse AML models driven by the KMT2A/MLLT3 fusion or by the constitutive coexpression of Hoxa9 and Meis1, both in vitro and in vivo. Loss of HERC1 enhanced nucleoside analog-induced cell death in both murine and human AML cell lines by compromising cell cycle progression. In-depth proteomic analysis and subsequent validation identified deoxycytidine kinase as a novel target of Herc1 in both mouse AML models. We observed that HERC1 is overexpressed in AML when compared with other cancer types and that higher HERC1 expression was associated with shorter overall survival in patients with AML in the The Cancer Gene Atlas program (TCGA) and BEAT-AML cohorts. Collectively, this study highlights the importance of HERC1 in the response of AML cells to nucleoside analogs, thereby establishing this E3 ubiquitin ligase as a novel predictive biomarker and potential therapeutic target for the treatment of AML.
© 2024 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for G.G.V. is Paraza Pharma Inc.
The current affiliation for M.J. is Paraza Pharma Inc.
The current affiliation for W.W.L.P. is Strand Therapeutics Inc.
Figures






References
-
- Dohner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140(12):1345–1377. - PubMed
-
- SEER. 2021.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
