

In vivo vulnerabilities to GPX4 and HDAC inhibitors in drug-persistent versus drug-resistant BRAFV600E lung adenocarcinoma
- PMID: 39094577
- PMCID: PMC11384943
- DOI: 10.1016/j.xcrm.2024.101663
In vivo vulnerabilities to GPX4 and HDAC inhibitors in drug-persistent versus drug-resistant BRAFV600E lung adenocarcinoma
Abstract
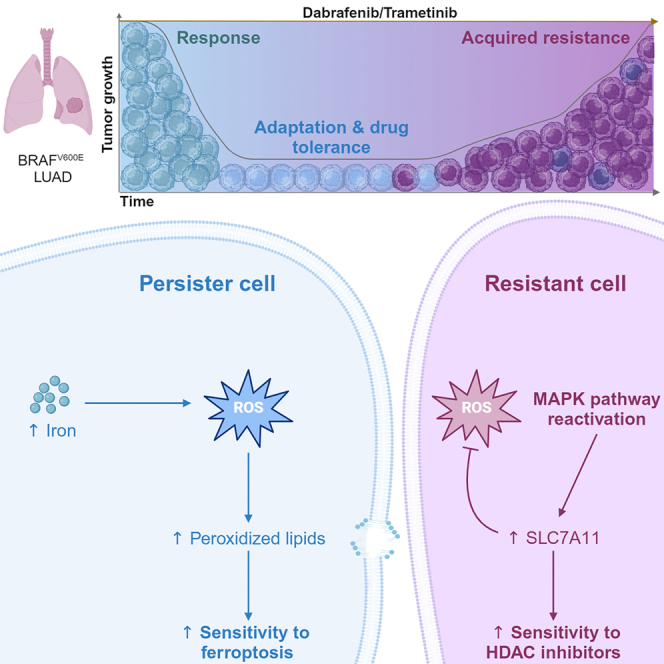
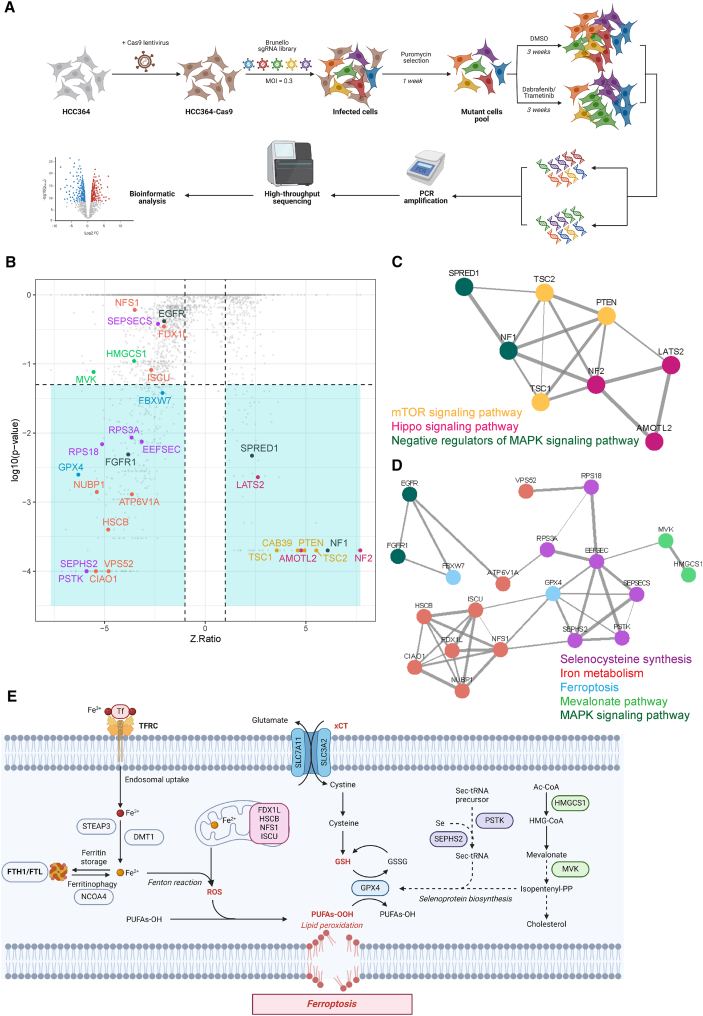
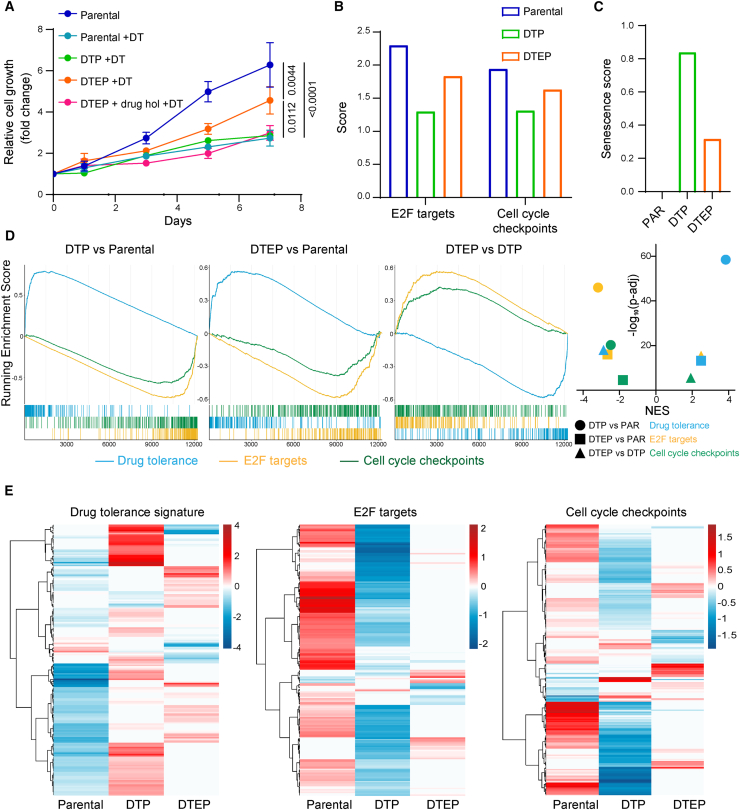
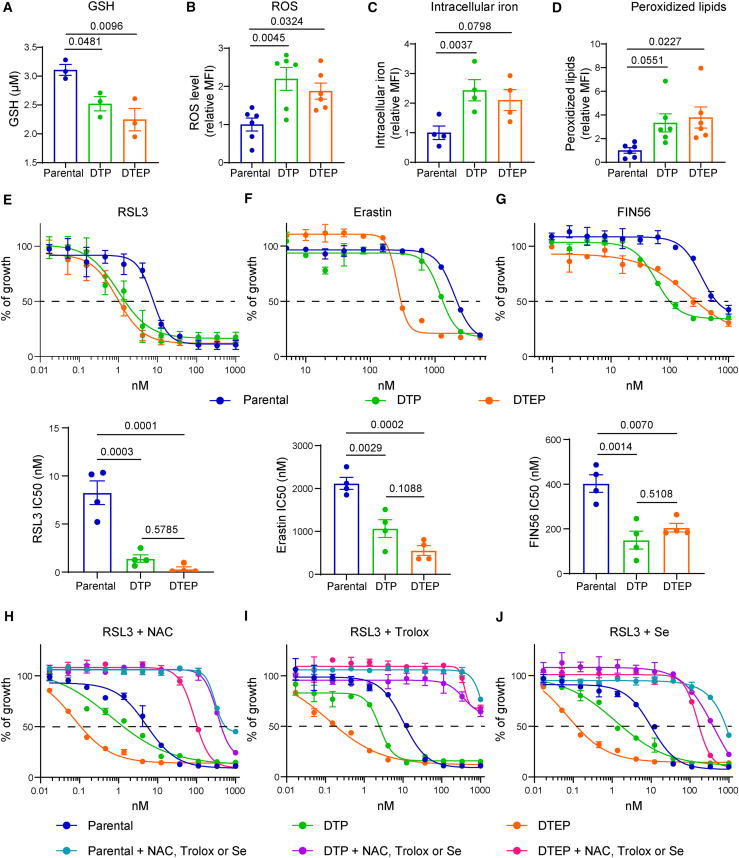
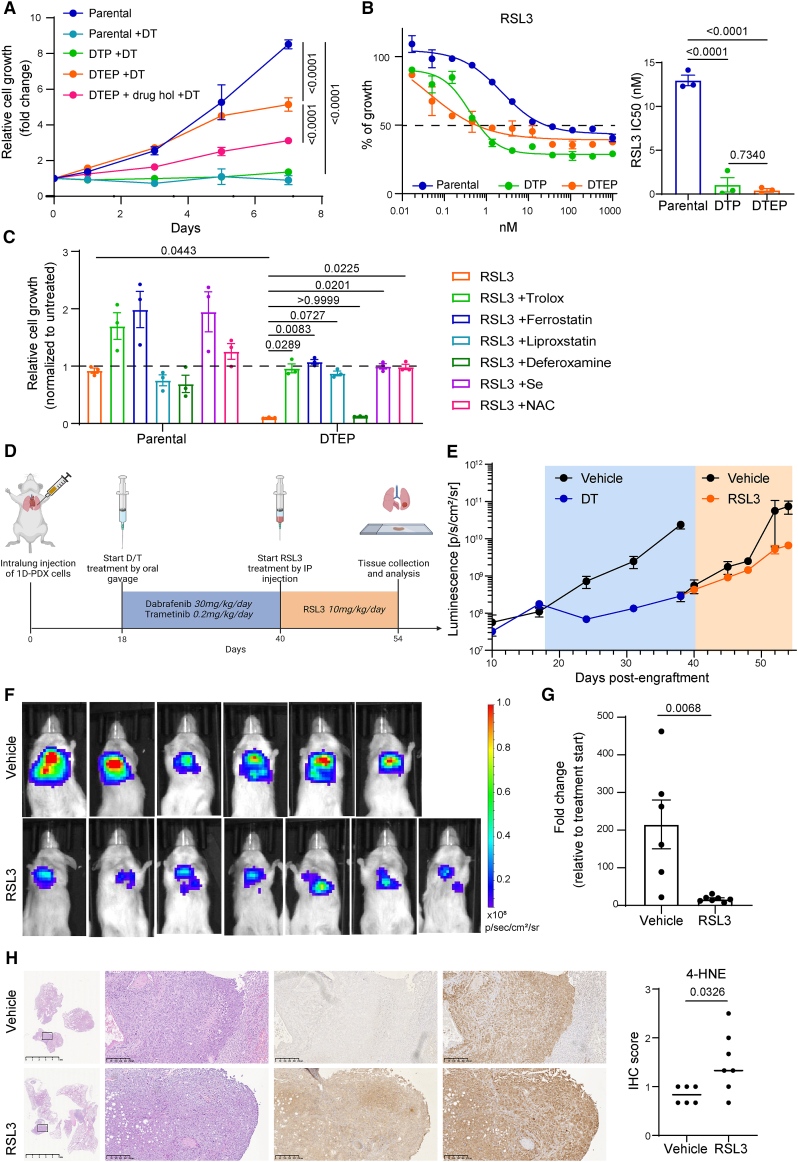
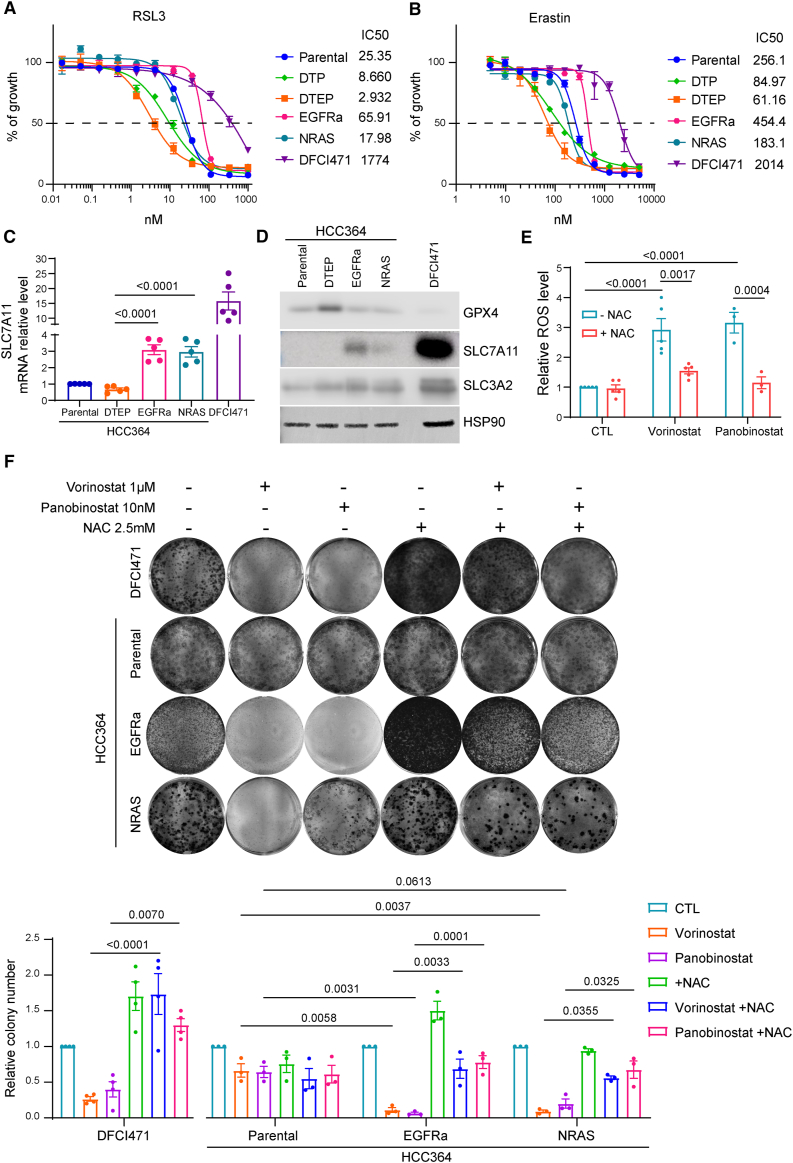
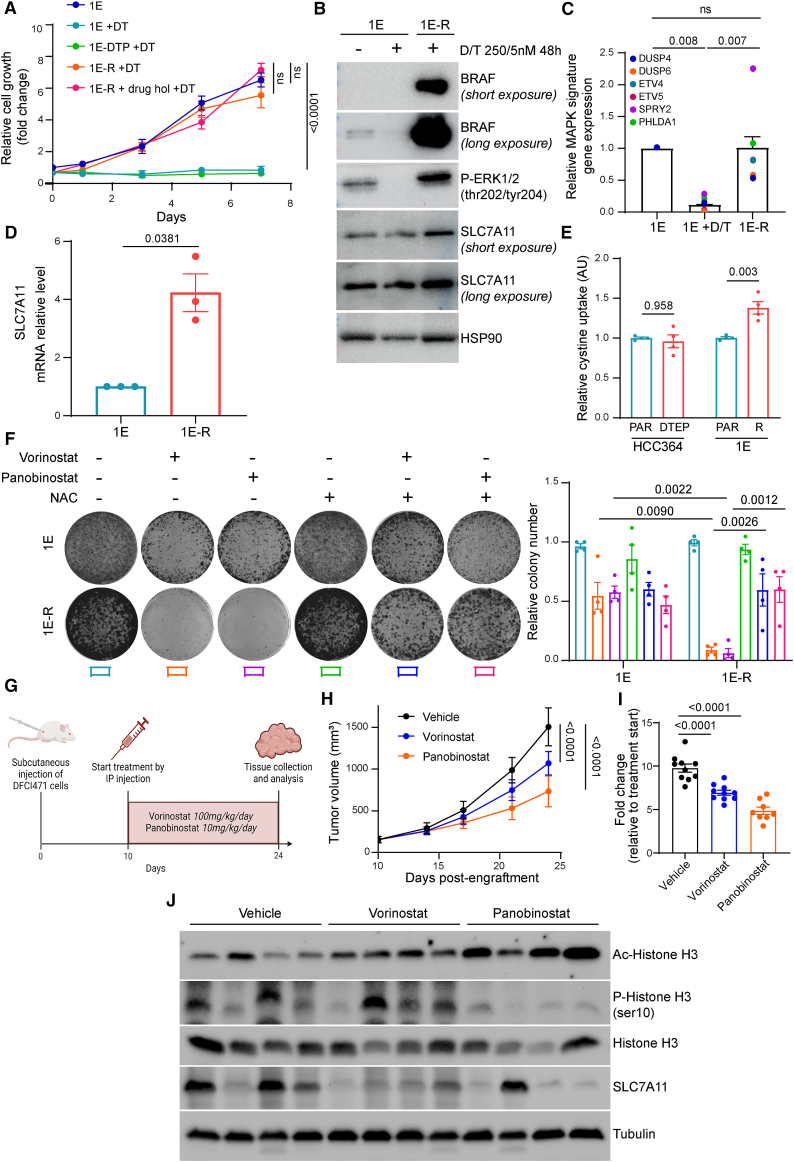
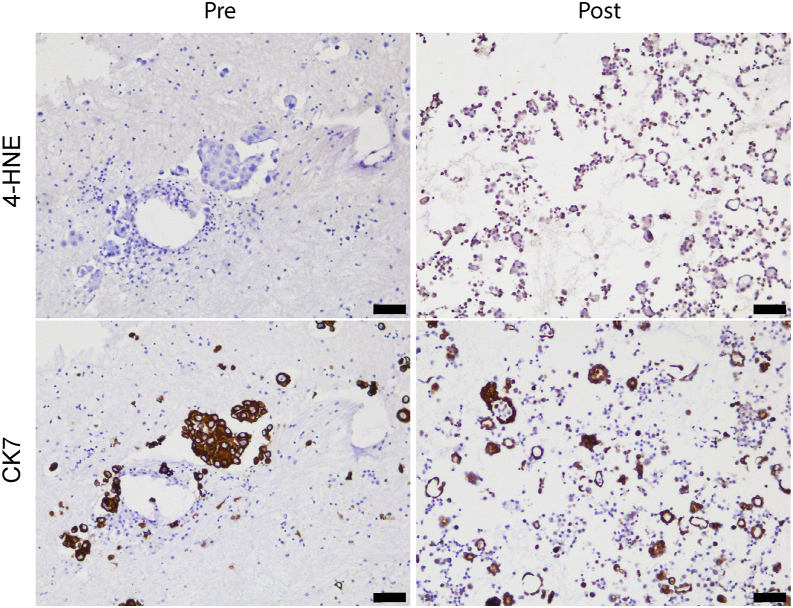
The current targeted therapy for BRAFV600E-mutant lung cancer consists of a dual blockade of RAF/MEK kinases often combining dabrafenib/trametinib (D/T). This regimen extends survival when compared to single-agent treatments, but disease progression is unavoidable. By using whole-genome CRISPR screening and RNA sequencing, we characterize the vulnerabilities of both persister and D/T-resistant cellular models. Oxidative stress together with concomitant induction of antioxidant responses is boosted by D/T treatment. However, the nature of the oxidative damage, the choice of redox detoxification systems, and the resulting therapeutic vulnerabilities display stage-specific differences. Persister cells suffer from lipid peroxidation and are sensitive to ferroptosis upon GPX4 inhibition in vivo. Biomarkers of lipid peroxidation are detected in clinical samples following D/T treatment. Acquired alterations leading to mitogen-activated protein kinase (MAPK) reactivation enhance cystine transport to boost GPX4-independent antioxidant responses. Similarly to BRAFV600E-mutant melanoma, histone deacetylase (HDAC) inhibitors decrease D/T-resistant cell viability and extend therapeutic response in vivo.
Keywords: BRAF oncogene; GPX4 inhibition; HDAC inhibition; ferroptosis; lung adenocarcinoma; persister cells; targeted therapy.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests D.S. received research fees from Aelin Therapeutics. C.A. received research fees from Revolution Medicines, Aelin Therapeutics, Verastem, Roche, and Boehringer Ingelheim. E.N. reports research funding from Pfizer and Roche. P.L. is listed as an inventor on patent applications filed by MSKCC that describe approaches to treat KRAS or BRAF-mutant tumors. P.A.J. has received consulting fees from AstraZeneca, Boehringer Ingelheim, Pfizer, Roche/Genentech, Takeda Oncology, ACEA Biosciences, Eli Lilly and Company, Araxes Pharma, Ignyta, Mirati Therapeutics, Novartis, Loxo Oncology, Daiichi Sankyo, Sanofi Oncology, Voronoi, SFJ Pharmaceuticals, Takeda Oncology, Transcenta, Silicon Therapeutics, Syndax, Nuvalent, Bayer, Eisai, Biocartis, Allorion Therapeutics, Accutar Biotech, Monte Rosa, Scorpion Therapeutics, Merus, Frontier Medicines, Hongyun Biotechnology, Duality, and AbbVie; post-marketing royalties from DFCI-owned intellectual property on EGFR mutations licensed to Lab Corp; sponsored research agreements with AstraZeneca, Daiichi Sankyo, Puma, Boehringer Ingelheim, Eli Lilly and Company, Revolution Medicines, and Astellas Pharmaceuticals; and stock ownership in Gatekeeper Pharmaceuticals. M.M.A. reports grants and personal fees from Genentech, grants and personal fees from Bristol Myers Squibb, personal fees from Merck, grants and personal fees from AstraZeneca, grants from Lilly, and personal fees from Maverick, Blueprint Medicine, Syndax, Ariad, Nektar, Gritstone, ArcherDX, Mirati, NextCure, Novartis, EMD Serono, and Panvaxal/NovaRx, outside the submitted work.
Figures
References
-
- Wiesweg M., Preuss C., Roeper J., Metzenmacher M., Eberhardt W., Stropiep U., Wedeken K., Reis H., Herold T., Darwiche K., et al. BRAF mutations and BRAF mutation functional class have no negative impact on the clinical outcome of advanced NSCLC and associate with susceptibility to immunotherapy. Eur. J. Cancer. 2021;149:211–221. doi: 10.1016/j.ejca.2021.02.036. - DOI - PubMed
-
- Dagogo-Jack I., Martinez P., Yeap B.Y., Ambrogio C., Ferris L.A., Lydon C., Nguyen T., Jessop N.A., Iafrate A.J., Johnson B.E., et al. Impact of BRAF Mutation Class on Disease Characteristics and Clinical Outcomes in BRAF-mutant Lung Cancer. Clin. Cancer Res. 2019;25:158–165. doi: 10.1158/1078-0432.CCR-18-2062. - DOI - PubMed
-
- Planchard D., Besse B., Groen H.J.M., Hashemi S.M.S., Mazieres J., Kim T.M., Quoix E., Souquet P.J., Barlesi F., Baik C., et al. Phase 2 Study of Dabrafenib Plus Trametinib in Patients With BRAF V600E-Mutant Metastatic NSCLC: Updated 5-Year Survival Rates and Genomic Analysis. J. Thorac. Oncol. 2022;17:103–115. doi: 10.1016/j.jtho.2021.08.011. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
