

Cancer, metastasis, and the epigenome
- PMID: 39095874
- PMCID: PMC11295362
- DOI: 10.1186/s12943-024-02069-w
Cancer, metastasis, and the epigenome
Abstract
Cancer is the second leading cause of death worldwide and disease burden is expected to increase globally throughout the next several decades, with the majority of cancer-related deaths occurring in metastatic disease. Cancers exhibit known hallmarks that endow them with increased survival and proliferative capacities, frequently as a result of de-stabilizing mutations. However, the genomic features that resolve metastatic clones from primary tumors are not yet well-characterized, as no mutational landscape has been identified as predictive of metastasis. Further, many cancers exhibit no known mutation signature. This suggests a larger role for non-mutational genome re-organization in promoting cancer evolution and dissemination. In this review, we highlight current critical needs for understanding cell state transitions and clonal selection advantages for metastatic cancer cells. We examine links between epigenetic states, genome structure, and misregulation of tumor suppressors and oncogenes, and discuss how recent technologies for understanding domain-scale regulation have been leveraged for a more complete picture of oncogenic and metastatic potential.
Keywords: Chromatin state; EMT; Epigenetic; Metastasis; Topology; Transcription.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
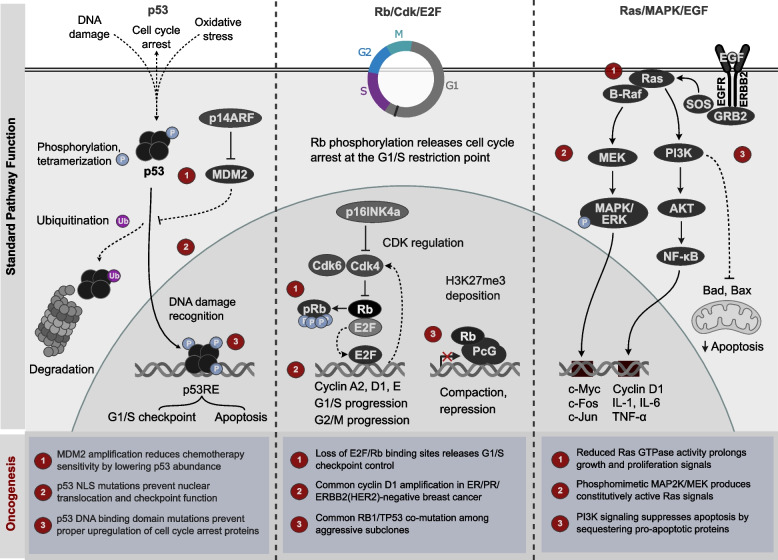
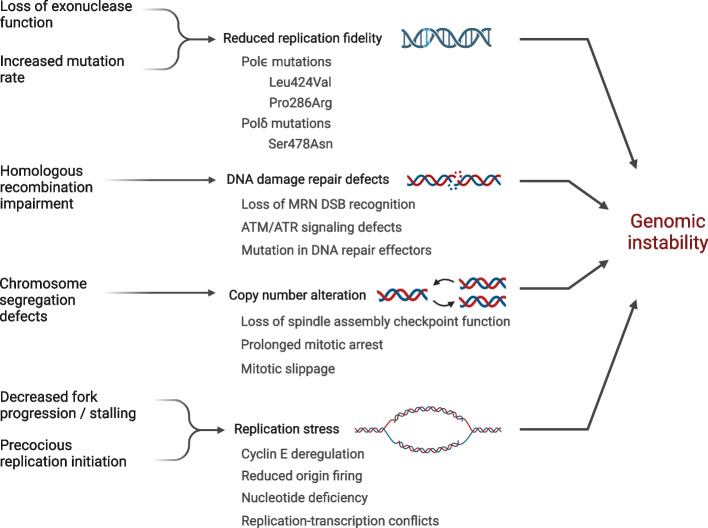
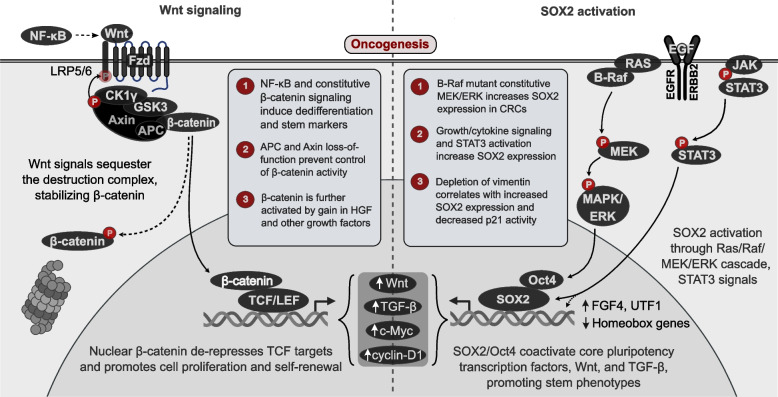
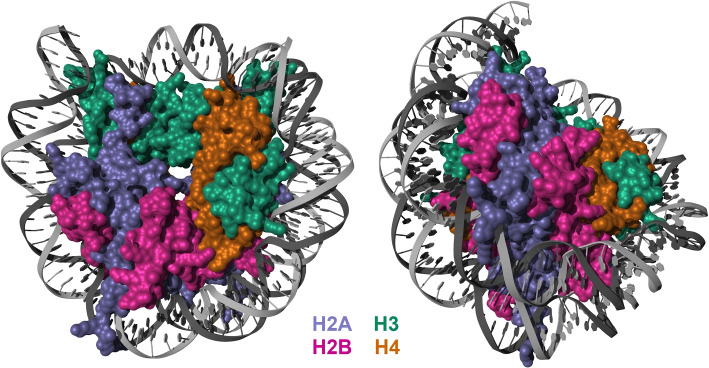
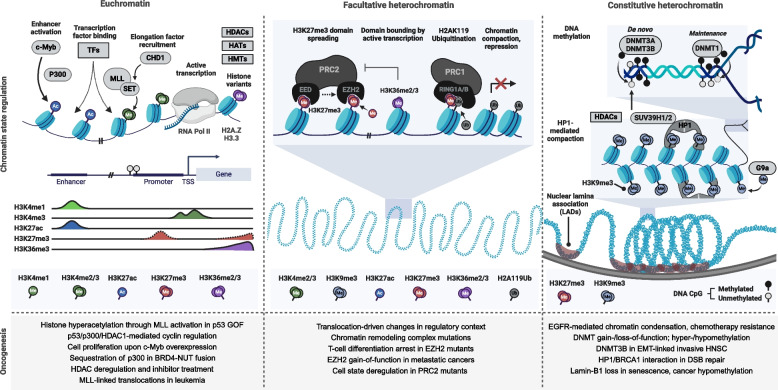
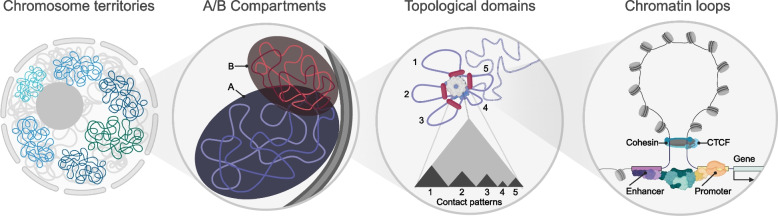
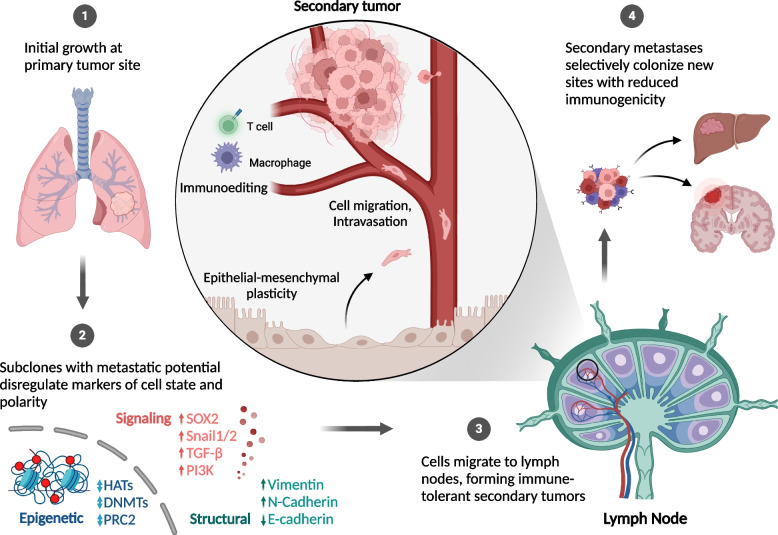
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
