

Protein-extending ACTN2 frameshift variants cause variable myopathy phenotypes by protein aggregation
- PMID: 39095936
- PMCID: PMC11537131
- DOI: 10.1002/acn3.52154
Protein-extending ACTN2 frameshift variants cause variable myopathy phenotypes by protein aggregation
Abstract
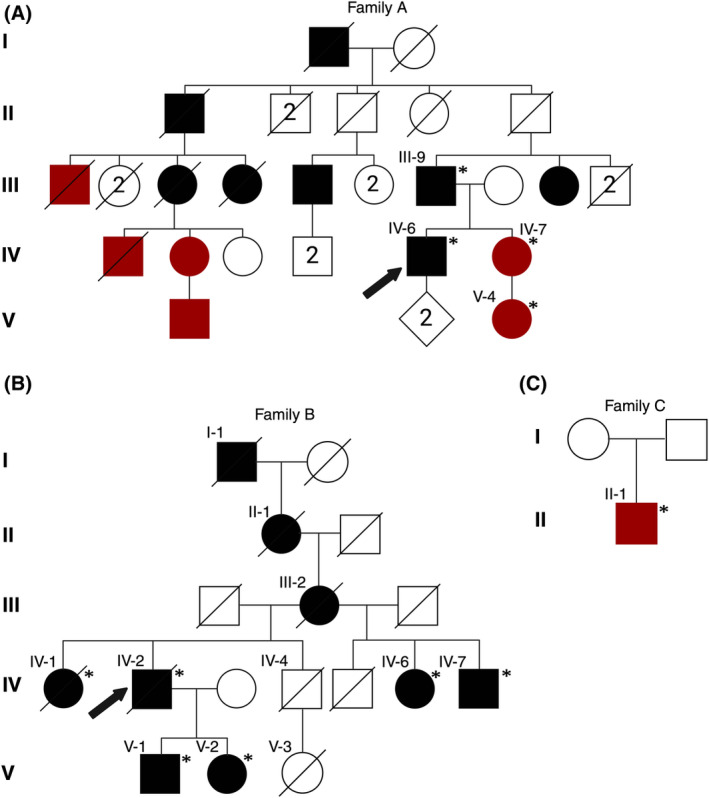
Objective: The objective of the study is to characterize the pathomechanisms underlying actininopathies. Distal myopathies are a group of rare, inherited muscular disorders characterized by progressive loss of muscle fibers that begin in the distal parts of arms and legs. Recently, variants in a new disease gene, ACTN2, have been shown to cause distal myopathy. ACTN2, a gene previously only associated with cardiomyopathies, encodes alpha-actinin-2, a protein expressed in both cardiac and skeletal sarcomeres. The primary function of alpha-actinin-2 is to link actin and titin to the sarcomere Z-disk. New ACTN2 variants are continuously discovered; however, the clinical significance of many variants remains unknown. Thus, lack of clear genotype-phenotype correlations in ACTN2-related diseases, actininopathies, persists.
Methods: Functional characterization in C2C12 cell model of several ACTN2 variants is conducted, including frameshift and missense variants associated with dominant and recessive actininopathies. We assess the genotype-phenotype correlations of actininopathies using clinical data from several patients carrying these variants.
Results: The results show that the missense variants associated with a recessive form of actininopathy do not cause detectable alpha-actinin-2 aggregates in the cell model. Conversely, dominant frameshift variants causing a protein extension do form alpha-actinin-2 aggregates.
Interpretation: The results suggest that alpha-actinin-2 aggregation is the disease mechanism underlying some dominant actininopathies, and thus, we recommend that protein-extending frameshift variants in ACTN2 should be classified as pathogenic. However, this mechanism is likely elicited by only a limited number of variants. Alternative functional characterization methods should be explored to further investigate other molecular mechanisms underlying actininopathies.
© 2024 The Author(s). Annals of Clinical and Translational Neurology published by Wiley Periodicals LLC on behalf of American Neurological Association.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures



References
-
- Beggs AH, Byers TJ, Knoll JH, Boyce FM, Bruns GA, Kunkel LM. Cloning and characterization of two human skeletal muscle alpha‐actinin genes located on chromosomes 1 and 11. J Biol Chem. 1992;267(13):9281‐9288. - PubMed
MeSH terms
Substances
Grants and funding
- DAF2019-199278/Chan Zuckerberg Initiative
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- Sigrid Jusélius Foundation
- U01 HG0011755/EY/NEI NIH HHS/United States
- R01 HG009141/HG/NHGRI NIH HHS/United States
- Folkhälsan Research Foundation
- 339437/Research Council of Finland
- Jane and Aatos Erkko Foundation
- Emil Aaltonen Foundation
- U01 HG011755/HG/NHGRI NIH HHS/United States
- Silicon Valley Community Foundation
- UM1 HG00890/HG/NHGRI NIH HHS/United States
- 23281/Association Francaise contre les Myopathies
- R01 HG00914/HL/NHLBI NIH HHS/United States
- Finska Läkaresällskapet
LinkOut - more resources
Full Text Sources