

PPARγ activation ameliorates cognitive impairment and chronic microglial activation in the aftermath of r-mTBI
- PMID: 39097742
- PMCID: PMC11297749
- DOI: 10.1186/s12974-024-03173-w
PPARγ activation ameliorates cognitive impairment and chronic microglial activation in the aftermath of r-mTBI
Abstract
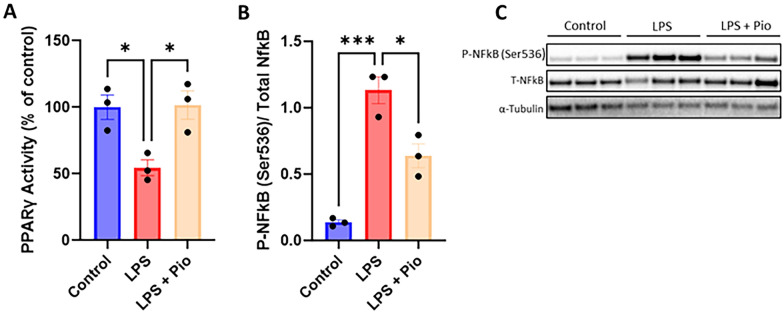
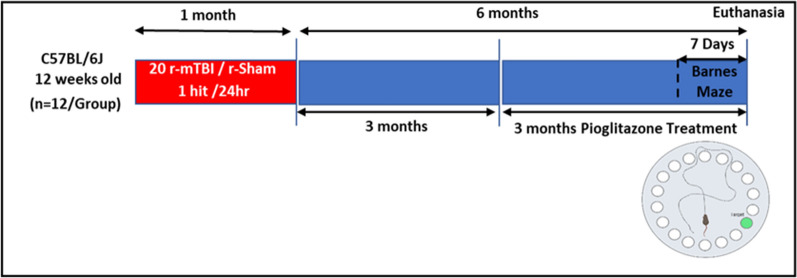
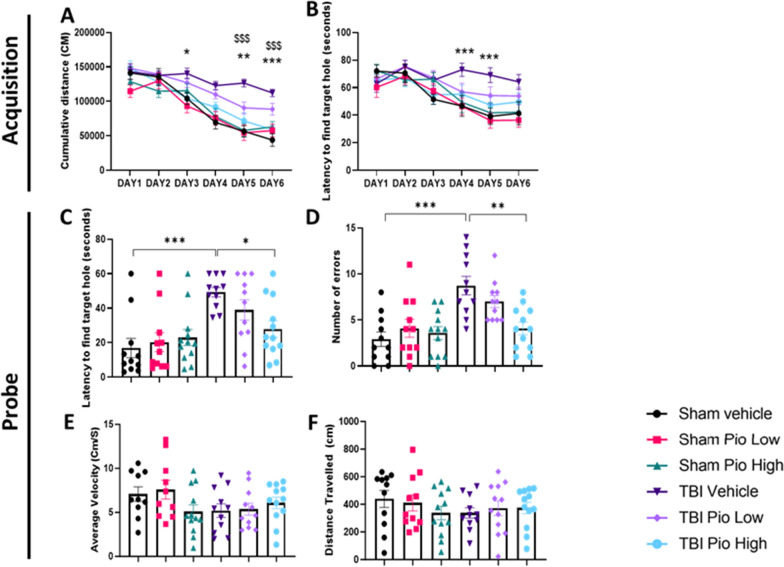
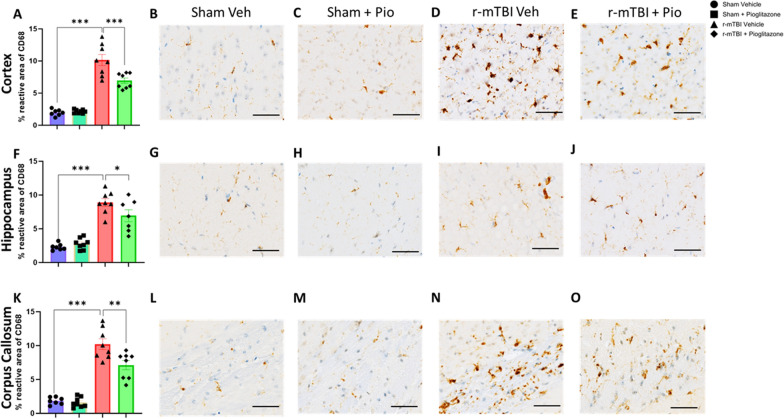
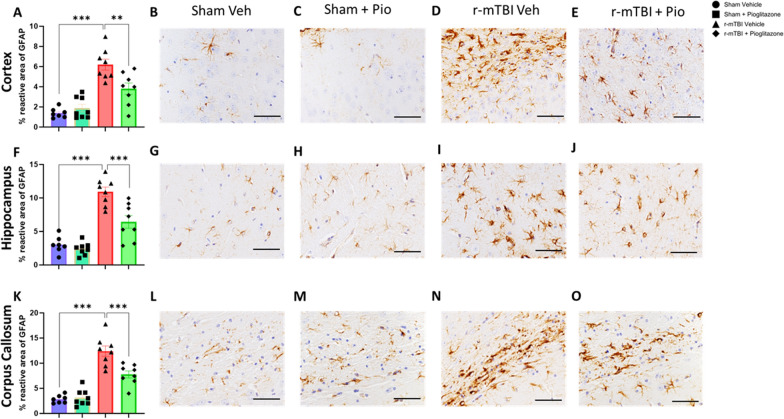
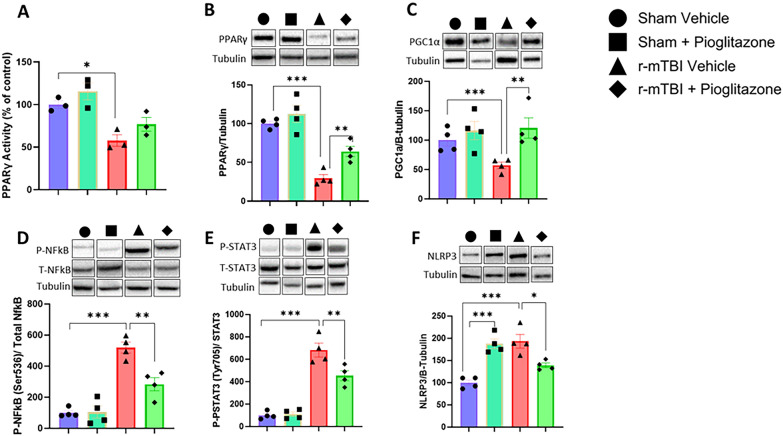
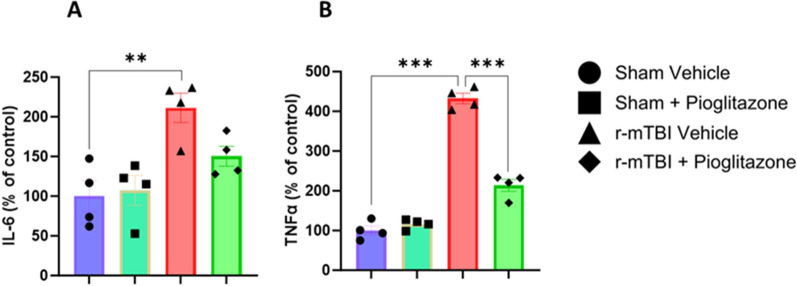
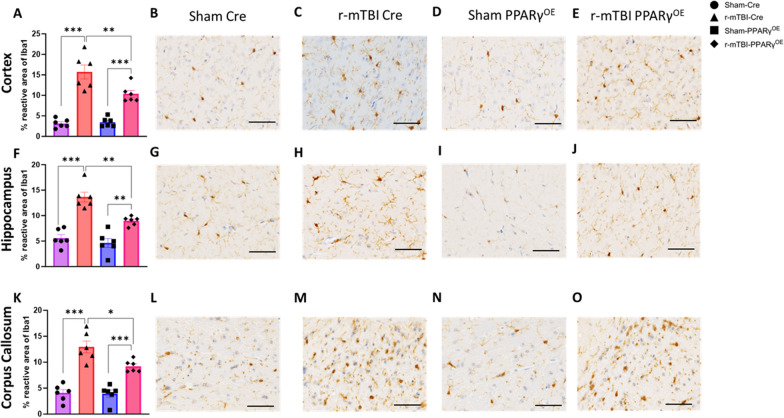
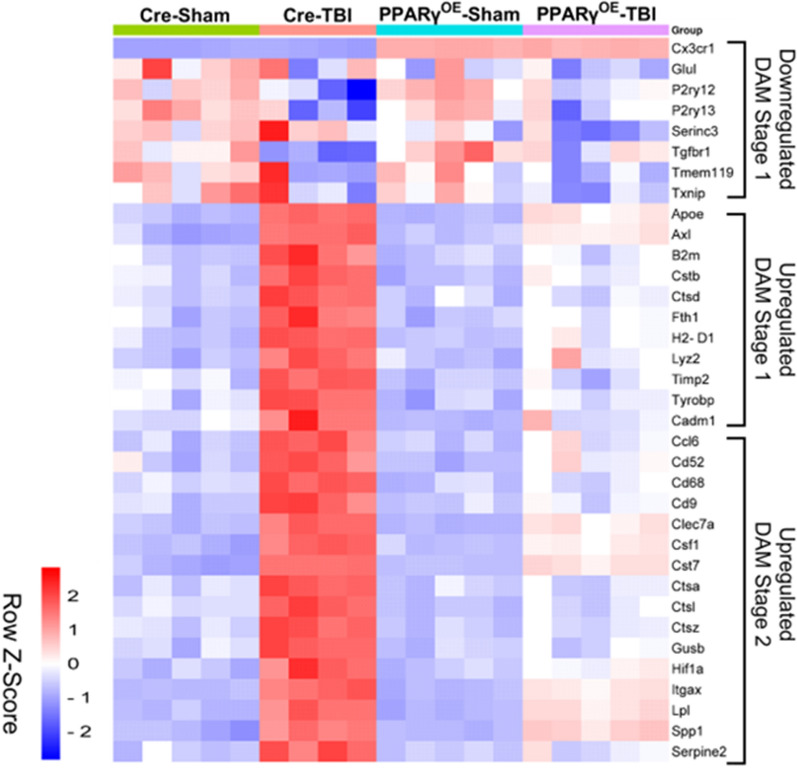
Chronic neuroinflammation and microglial activation are key mediators of the secondary injury cascades and cognitive impairment that follow exposure to repetitive mild traumatic brain injury (r-mTBI). Peroxisome proliferator-activated receptor-γ (PPARγ) is expressed on microglia and brain resident myeloid cell types and their signaling plays a major anti-inflammatory role in modulating microglial responses. At chronic timepoints following injury, constitutive PPARγ signaling is thought to be dysregulated, thus releasing the inhibitory brakes on chronically activated microglia. Increasing evidence suggests that thiazolidinediones (TZDs), a class of compounds approved from the treatment of diabetes mellitus, effectively reduce neuroinflammation and chronic microglial activation by activating the peroxisome proliferator-activated receptor-γ (PPARγ). The present study used a closed-head r-mTBI model to investigate the influence of the TZD Pioglitazone on cognitive function and neuroinflammation in the aftermath of r-mTBI exposure. We revealed that Pioglitazone treatment attenuated spatial learning and memory impairments at 6 months post-injury and reduced the expression of reactive microglia and astrocyte markers in the cortex, hippocampus, and corpus callosum. We then examined whether Pioglitazone treatment altered inflammatory signaling mechanisms in isolated microglia and confirmed downregulation of proinflammatory transcription factors and cytokine levels. To further investigate microglial-specific mechanisms underlying PPARγ-mediated neuroprotection, we generated a novel tamoxifen-inducible microglial-specific PPARγ overexpression mouse line and examined its influence on microglial phenotype following injury. Using RNA sequencing, we revealed that PPARγ overexpression ameliorates microglial activation, promotes the activation of pathways associated with wound healing and tissue repair (such as: IL10, IL4 and NGF pathways), and inhibits the adoption of a disease-associated microglia-like (DAM-like) phenotype. This study provides insight into the role of PPARγ as a critical regulator of the neuroinflammatory cascade that follows r-mTBI in mice and demonstrates that the use of PPARγ agonists such as Pioglitazone and newer generation TZDs hold strong therapeutic potential to prevent the chronic neurodegenerative sequelae of r-mTBI.
© 2024. The Author(s).
Conflict of interest statement
The authors declare they have no competing interests.
Figures





References
-
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Grüning BA, Guerler A, Hillman-Jackson J, Hiltemann S, Jalili V, Rasche H, Soranzo N, Goecks J, Taylor J, Nekrutenko A, Blankenberg D. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46(W1):W537–44. 10.1093/nar/gky379. 10.1093/nar/gky379 - DOI - PMC - PubMed
-
- Andrews S. FastQC: A quality control tool for High-Throughput sequence data. 2010. http://www.bioinformatics.babraham.ac.uk/Projects/Fastqc/. Accessed 19 Nov 2023.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
