

Geometric deep learning of protein-DNA binding specificity
- PMID: 39103447
- PMCID: PMC11399107
- DOI: 10.1038/s41592-024-02372-w
Geometric deep learning of protein-DNA binding specificity
Abstract
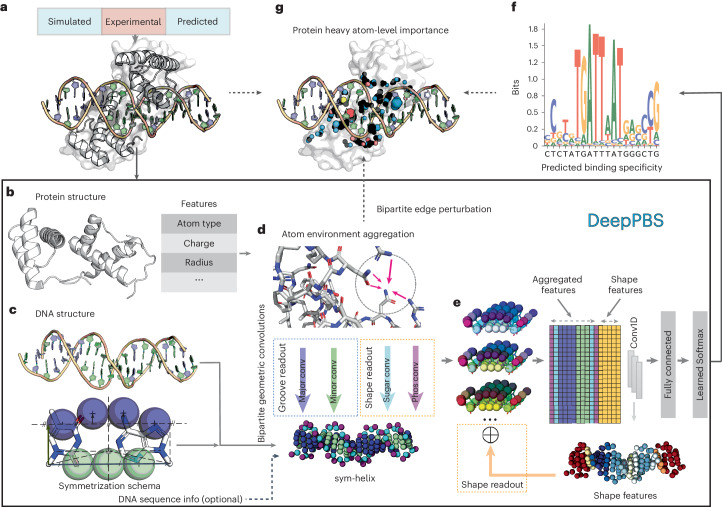
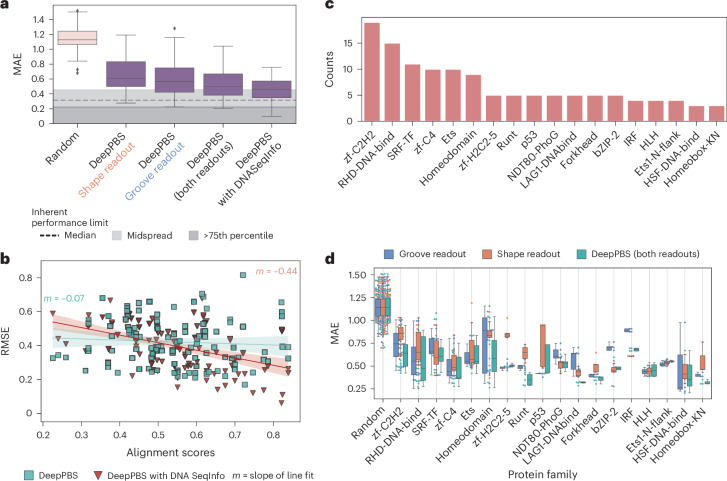
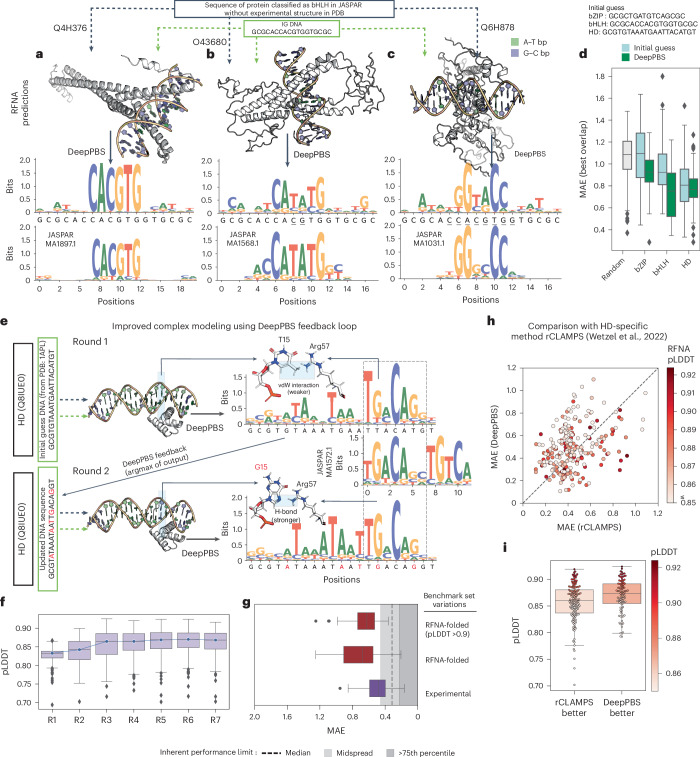
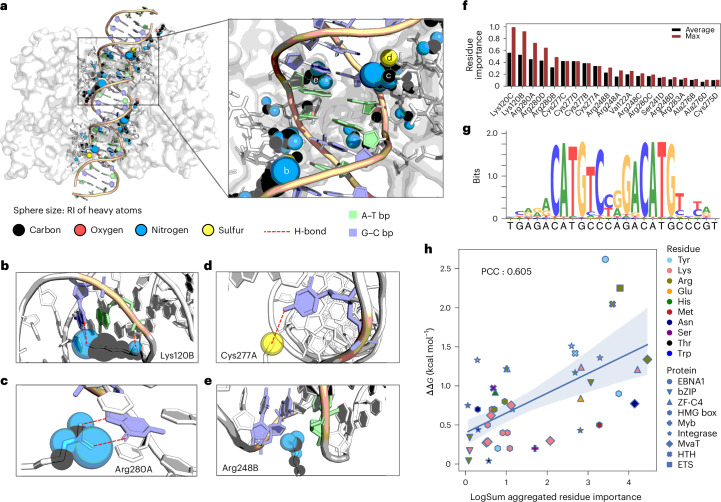
Predicting protein-DNA binding specificity is a challenging yet essential task for understanding gene regulation. Protein-DNA complexes usually exhibit binding to a selected DNA target site, whereas a protein binds, with varying degrees of binding specificity, to a wide range of DNA sequences. This information is not directly accessible in a single structure. Here, to access this information, we present Deep Predictor of Binding Specificity (DeepPBS), a geometric deep-learning model designed to predict binding specificity from protein-DNA structure. DeepPBS can be applied to experimental or predicted structures. Interpretable protein heavy atom importance scores for interface residues can be extracted. When aggregated at the protein residue level, these scores are validated through mutagenesis experiments. Applied to designed proteins targeting specific DNA sequences, DeepPBS was demonstrated to predict experimentally measured binding specificity. DeepPBS offers a foundation for machine-aided studies that advance our understanding of molecular interactions and guide experimental designs and synthetic biology.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Update of
-
DeepPBS: Geometric deep learning for interpretable prediction of protein-DNA binding specificity.bioRxiv [Preprint]. 2023 Dec 16:2023.12.15.571942. doi: 10.1101/2023.12.15.571942. bioRxiv. 2023. Update in: Nat Methods. 2024 Sep;21(9):1674-1683. doi: 10.1038/s41592-024-02372-w. PMID: 38293168 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
