

Bayesian reweighting of biomolecular structural ensembles using heterogeneous cryo-EM maps with the cryoENsemble method
- PMID: 39103467
- PMCID: PMC11300795
- DOI: 10.1038/s41598-024-68468-7
Bayesian reweighting of biomolecular structural ensembles using heterogeneous cryo-EM maps with the cryoENsemble method
Abstract
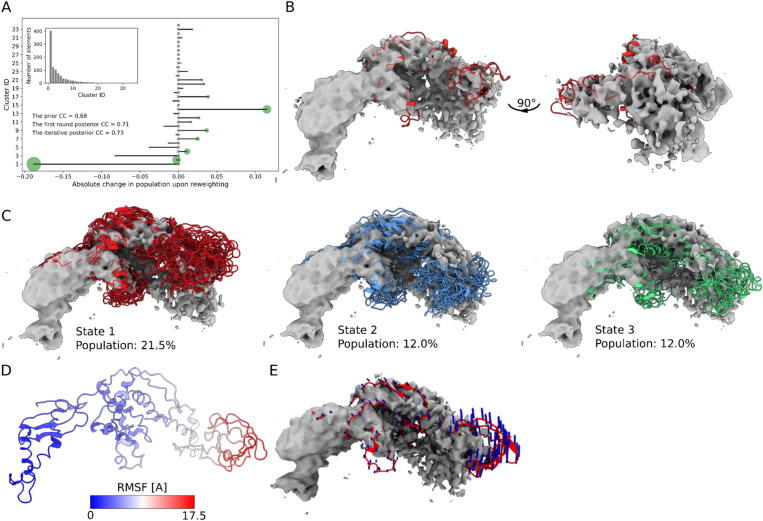
Cryogenic electron microscopy (cryo-EM) has emerged as a powerful method for the determination of structures of complex biological molecules. The accurate characterisation of the dynamics of such systems, however, remains a challenge. To address this problem, we introduce cryoENsemble, a method that applies Bayesian reweighting to conformational ensembles derived from molecular dynamics simulations to improve their agreement with cryo-EM data, thus enabling the extraction of dynamics information. We illustrate the use of cryoENsemble to determine the dynamics of the ribosome-bound state of the co-translational chaperone trigger factor (TF). We also show that cryoENsemble can assist with the interpretation of low-resolution, noisy or unaccounted regions of cryo-EM maps. Notably, we are able to link an unaccounted part of the cryo-EM map to the presence of another protein (methionine aminopeptidase, or MetAP), rather than to the dynamics of TF, and model its TF-bound state. Based on these results, we anticipate that cryoENsemble will find use for challenging heterogeneous cryo-EM maps for biomolecular systems encompassing dynamic components.
Keywords: Cryo-EM; Molecular dynamics simulations; Statistical inference; Structural biology; Trigger factor.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
