

Comprehensive analysis of sialylation-related genes and construct the prognostic model in sepsis
- PMID: 39103477
- PMCID: PMC11300640
- DOI: 10.1038/s41598-024-69185-x
Comprehensive analysis of sialylation-related genes and construct the prognostic model in sepsis
Abstract
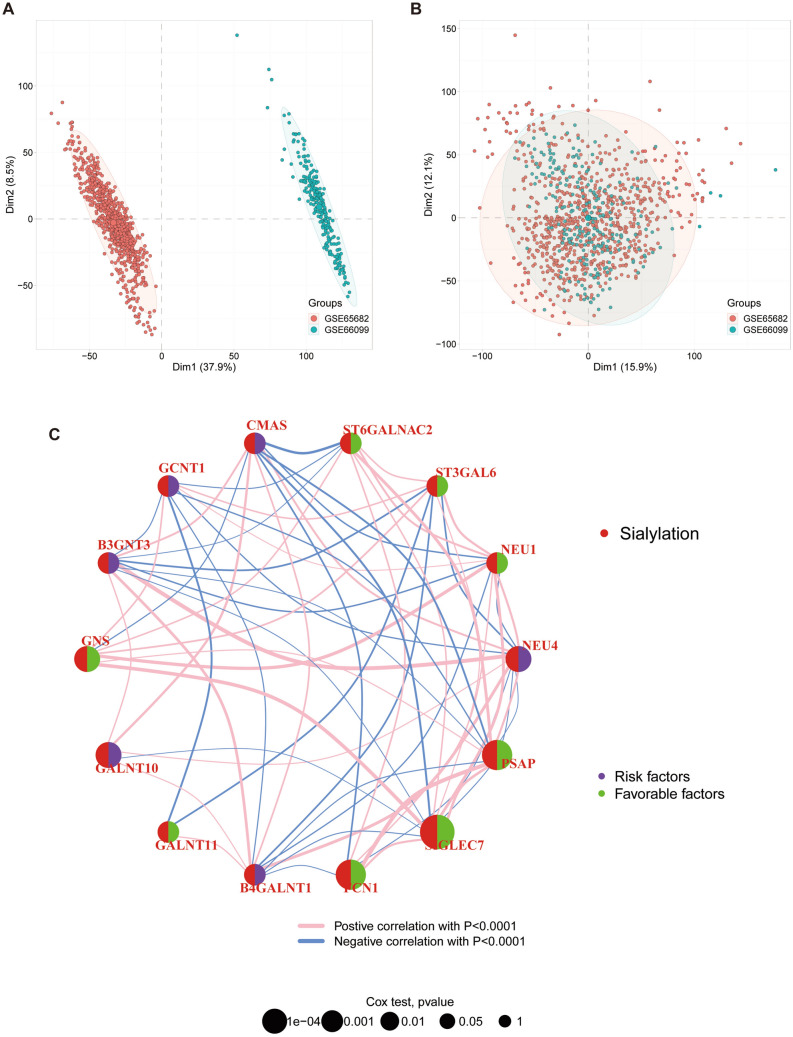
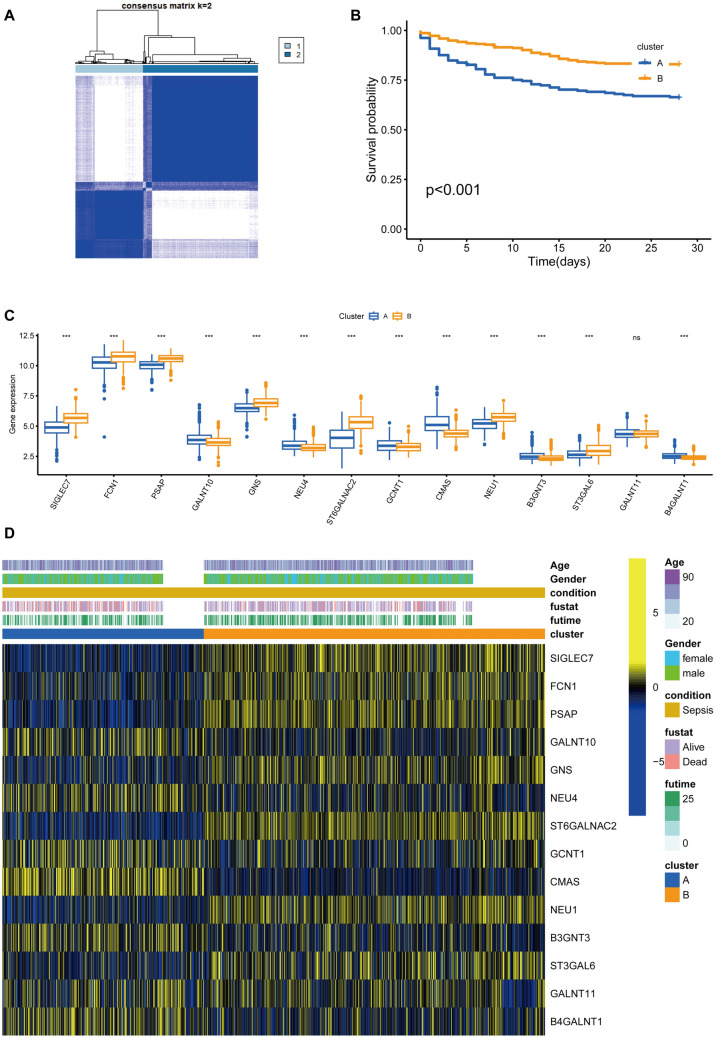
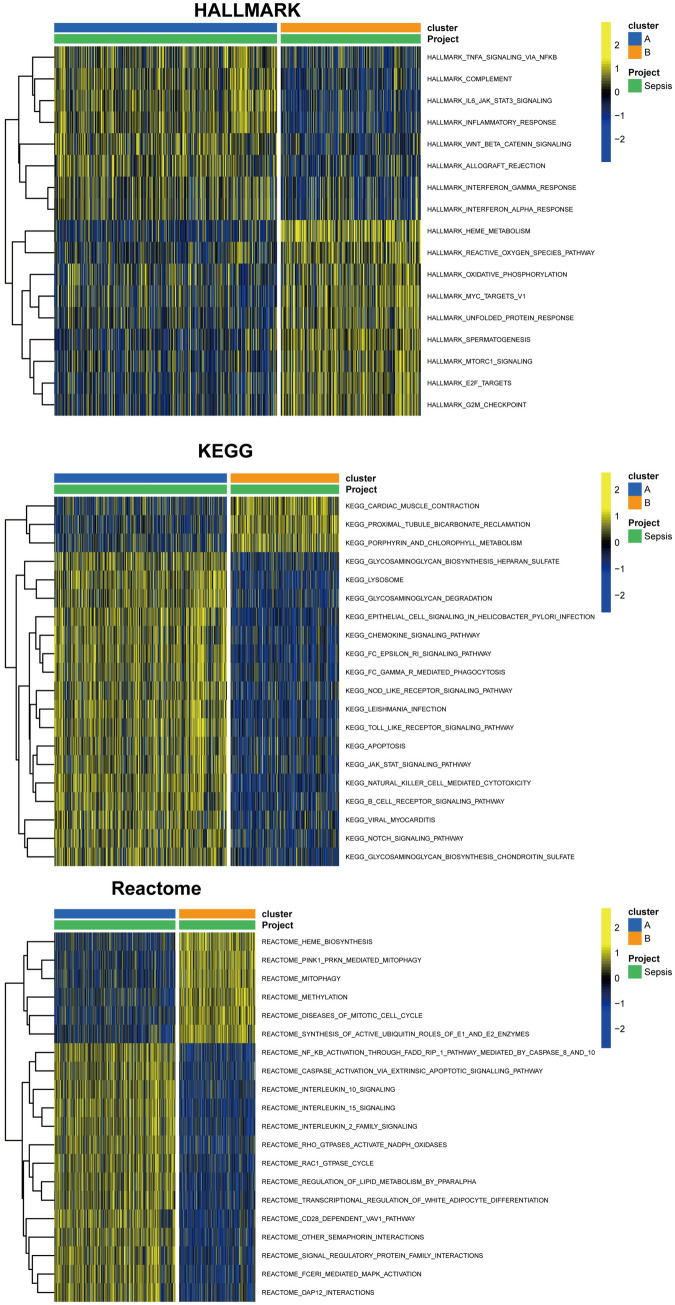
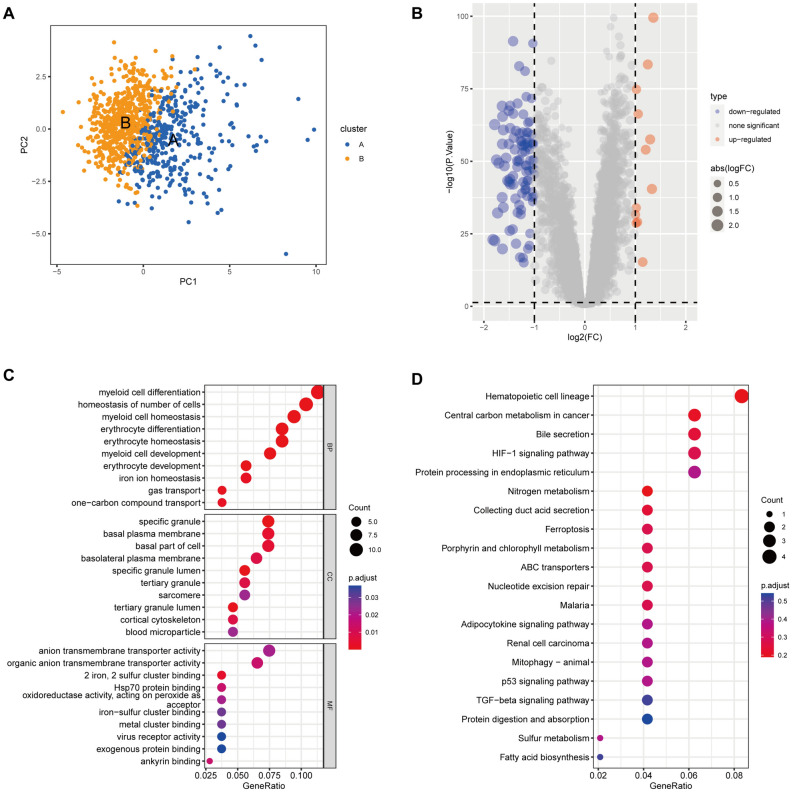
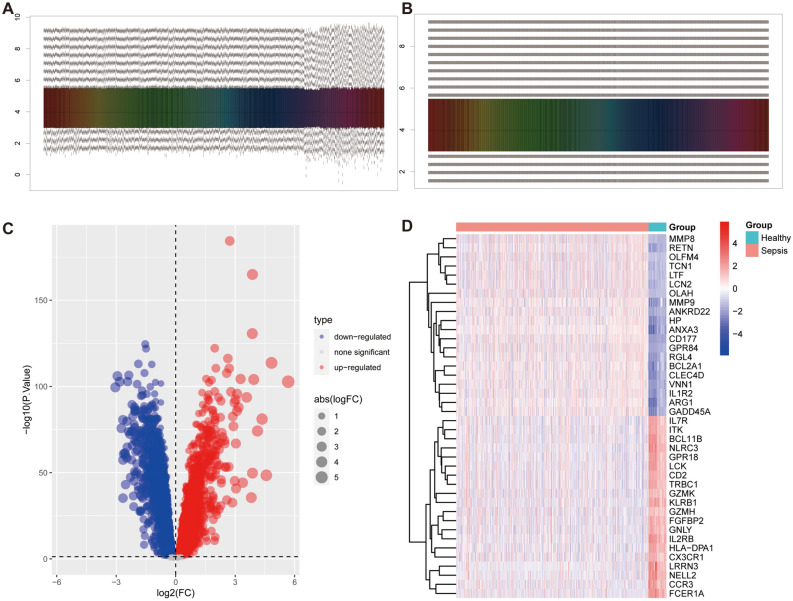
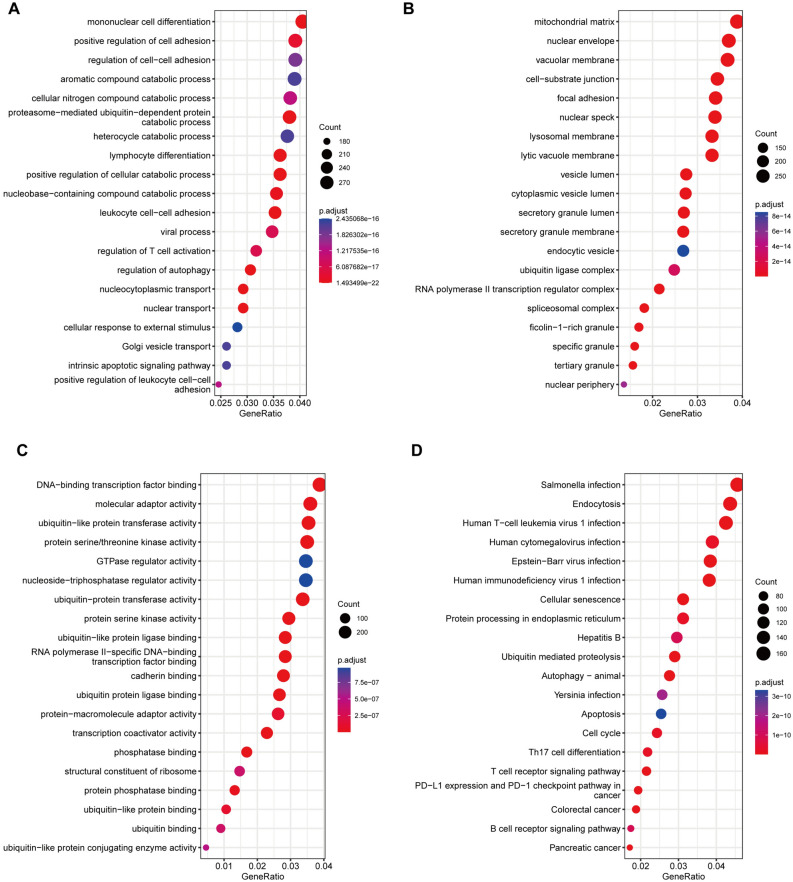
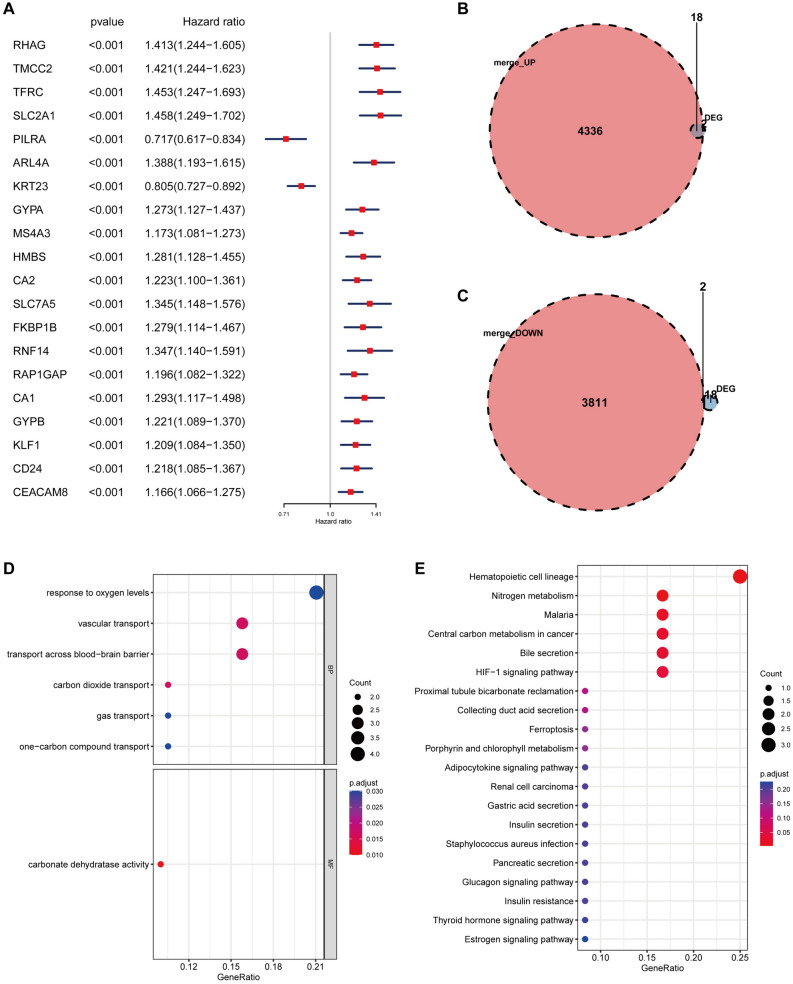
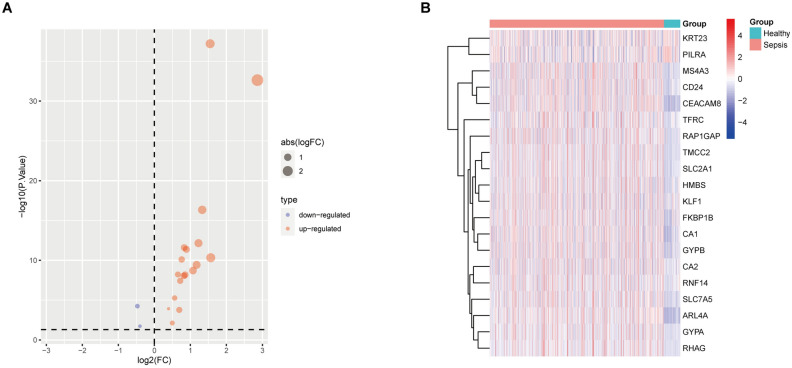
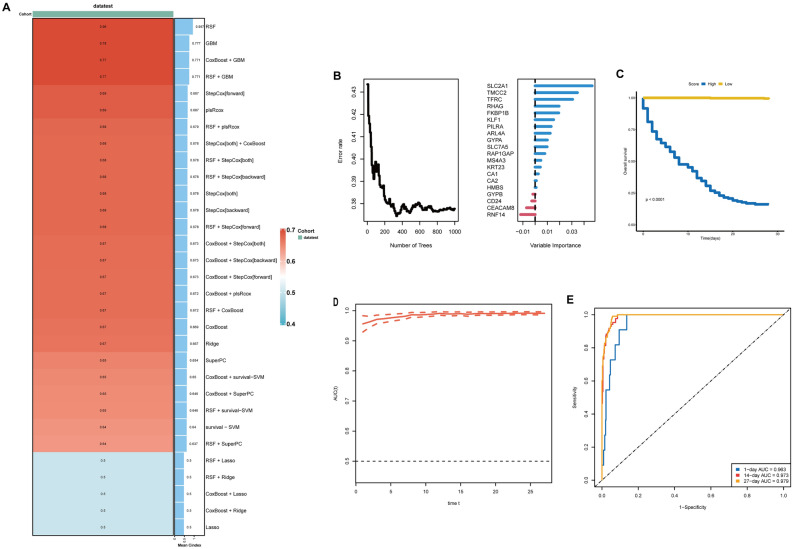
Sepsis, a life-threatening syndrome, continues to be a significant public health issue worldwide. Sialylation is a hot potential marker that affects the surface of a variety of cells. However, the role of genes related to sialylation and sepsis has not been fully explored. Bulk RNA-seq data sets (GSE66099 and GSE65682) were obtained from the open-access databases GEO. The classification of sepsis samples into subtypes was achieved by employing the R package "ConsensusClusterPlus" on the bulk RNA-seq data. Hub genes were discerned through the application of the R package "limma" and univariate regression analysis, with the calculation of risk scores carried out using the R package "survminer". To identify the best learning method and construct a prognostic model, we used 21 different combinations of machine learning, and C-index ranking results of these combinations have been showed. ROC curves, time-dependent ROC curves, and Kaplan-Meier curves were utilized to evaluate the diagnostic accuracy of the model. The R packages "ESTIMATE" and "GSVA" were employed to quantify the fractions of immune cell infiltration in each sample. The bulk RNA-seq samples were categorized into two distinct sepsis subtypes utilizing 14 prognosis-related sialylation genes. A total of 20 differentially expressed genes (DEGs) were identified as being associated with the relationship between sepsis and sialylation. The RSF was used to identify key genes with importance scores higher than 0.01. The nine hub genes (SLA2A1, TMCC2, TFRC, RHAG, FKBP1B, KLF1, PILRA, ARL4A, and GYPA) with the importance values greater than 0.01 was selected for constructing the prognostic model. This research offers some understanding of the relationship between sepsis and sialylation. Besides, it contains one predictive model that might develop into diagnostic biomarkers for sepsis.
Keywords: Bulk RNA-seq; Sepsis; Sialylation.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
