

Genetic risk factors for COVID-19 and influenza are largely distinct
- PMID: 39103650
- PMCID: PMC11319199
- DOI: 10.1038/s41588-024-01844-1
Genetic risk factors for COVID-19 and influenza are largely distinct
Abstract
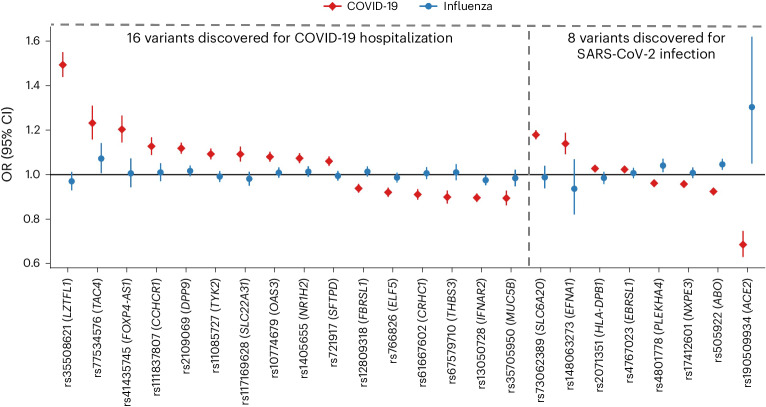
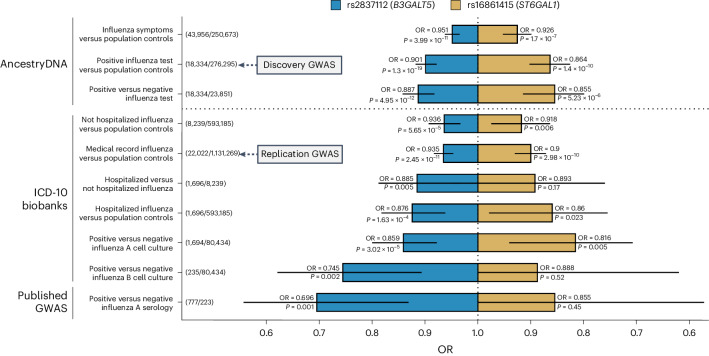
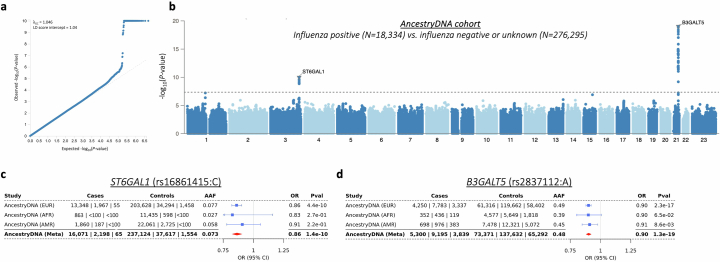
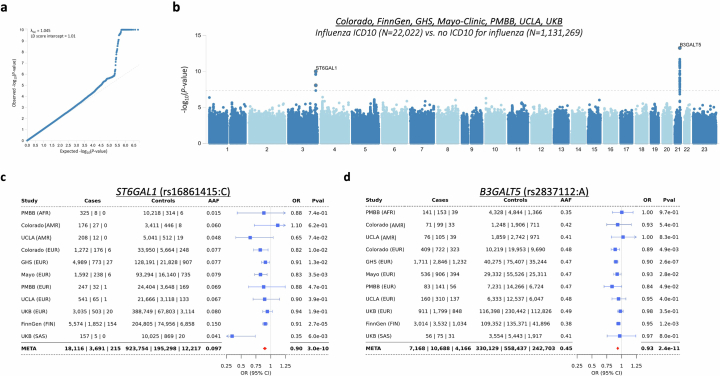
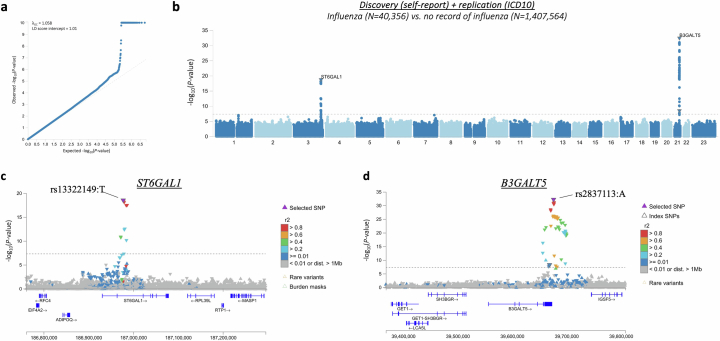
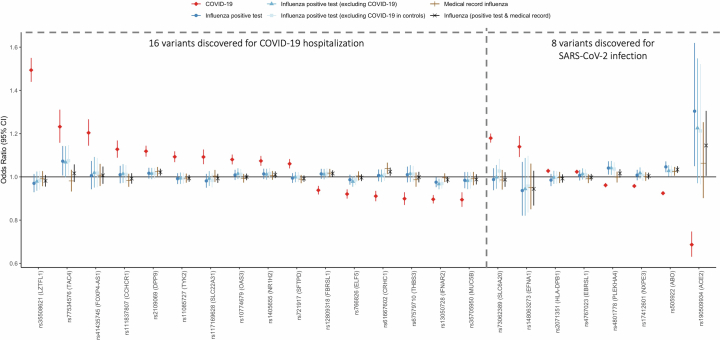
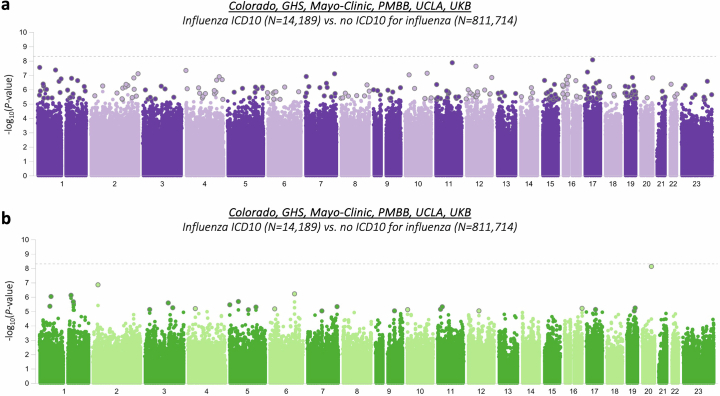
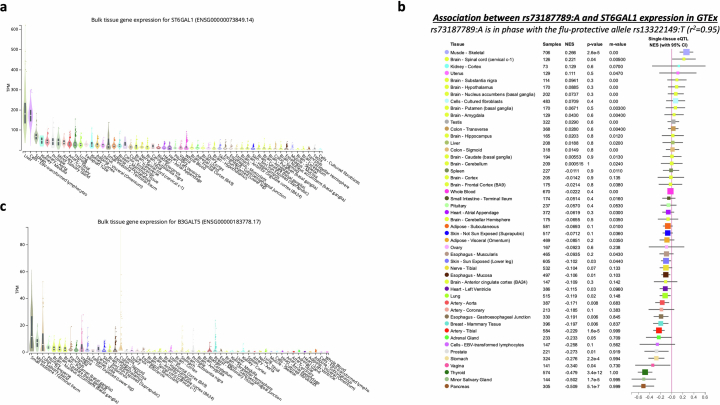
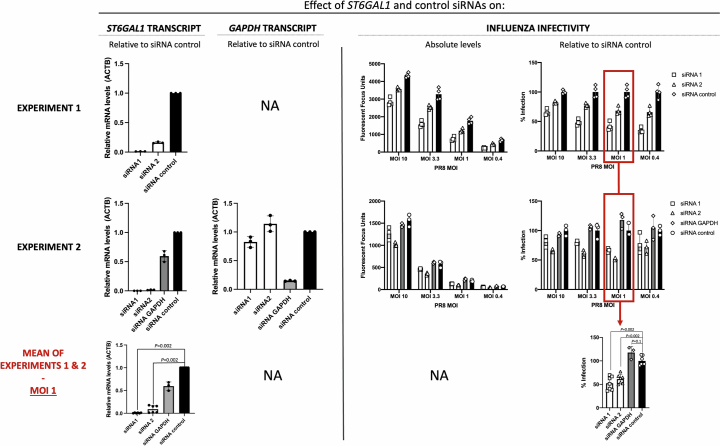
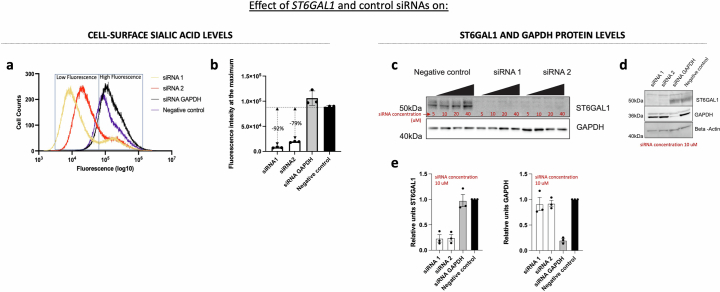
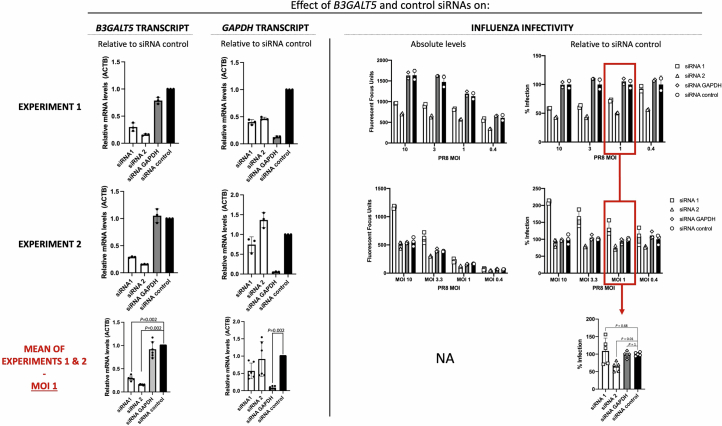
Coronavirus disease 2019 (COVID-19) and influenza are respiratory illnesses caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and influenza viruses, respectively. Both diseases share symptoms and clinical risk factors1, but the extent to which these conditions have a common genetic etiology is unknown. This is partly because host genetic risk factors are well characterized for COVID-19 but not for influenza, with the largest published genome-wide association studies for these conditions including >2 million individuals2 and about 1,000 individuals3-6, respectively. Shared genetic risk factors could point to targets to prevent or treat both infections. Through a genetic study of 18,334 cases with a positive test for influenza and 276,295 controls, we show that published COVID-19 risk variants are not associated with influenza. Furthermore, we discovered and replicated an association between influenza infection and noncoding variants in B3GALT5 and ST6GAL1, neither of which was associated with COVID-19. In vitro small interfering RNA knockdown of ST6GAL1-an enzyme that adds sialic acid to the cell surface, which is used for viral entry-reduced influenza infectivity by 57%. These results mirror the observation that variants that downregulate ACE2, the SARS-CoV-2 receptor, protect against COVID-19 (ref. 7). Collectively, these findings highlight downregulation of key cell surface receptors used for viral entry as treatment opportunities to prevent COVID-19 and influenza.
© 2024. The Author(s).
Conflict of interest statement
J.A.K., A.M., D.S., S.A.D.G., S.B., X.-M.Y., G.T., H.M., C.S., M.D.K., J.E.H., N.B., R.L., E.M., X.B., A.J.M., J. Mbatchou, K.W., W.J.S., A.R.S., J. Marchini, J.D.O., L.H., J.G.R., A.E., C.K., K.K., A. Baum, M.N.C., K.S., A. Baras, G.R.A. and M.A.R.F. are current employees or stockholders of Regeneron Genetics Center or Regeneron Pharmaceuticals. G.H.L.R., M.V.C., D.S.P., S.C.K., H.G., A. Baltzell, A.R.G., S.R.M., R.P., D.A.T., M.Z., K.A.R., E.L.H. and C.A.B. are current or past employees of AncestryDNA and may hold equity in AncestryDNA. The other authors declare no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
