

BRCA1 secondary splice-site mutations drive exon-skipping and PARP inhibitor resistance
- PMID: 39103848
- PMCID: PMC11299415
- DOI: 10.1186/s12943-024-02048-1
BRCA1 secondary splice-site mutations drive exon-skipping and PARP inhibitor resistance
Abstract
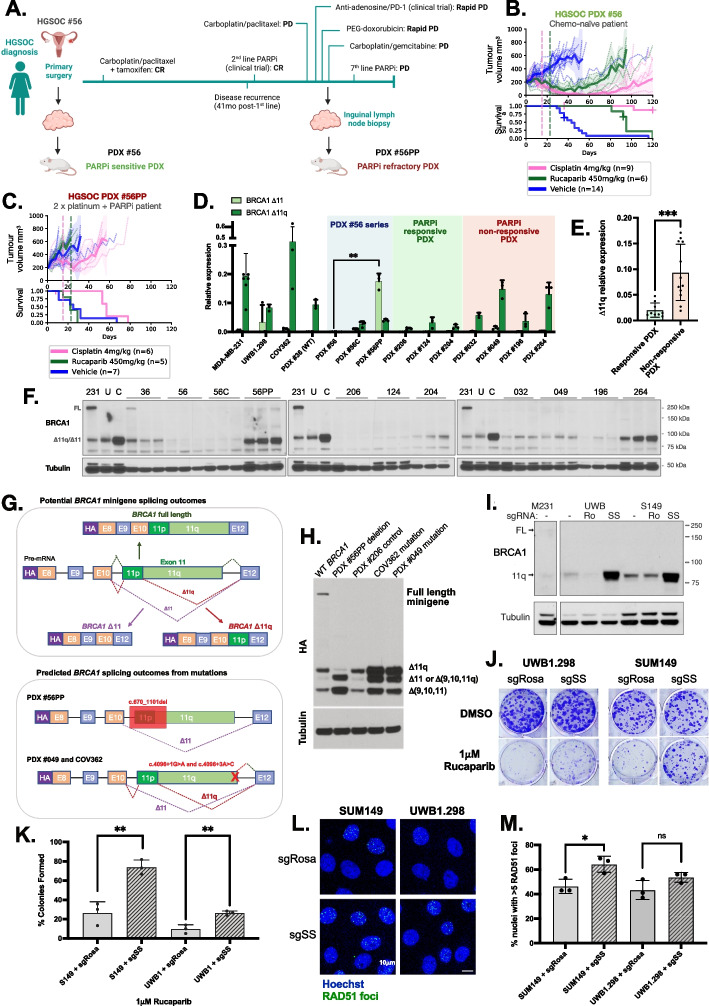
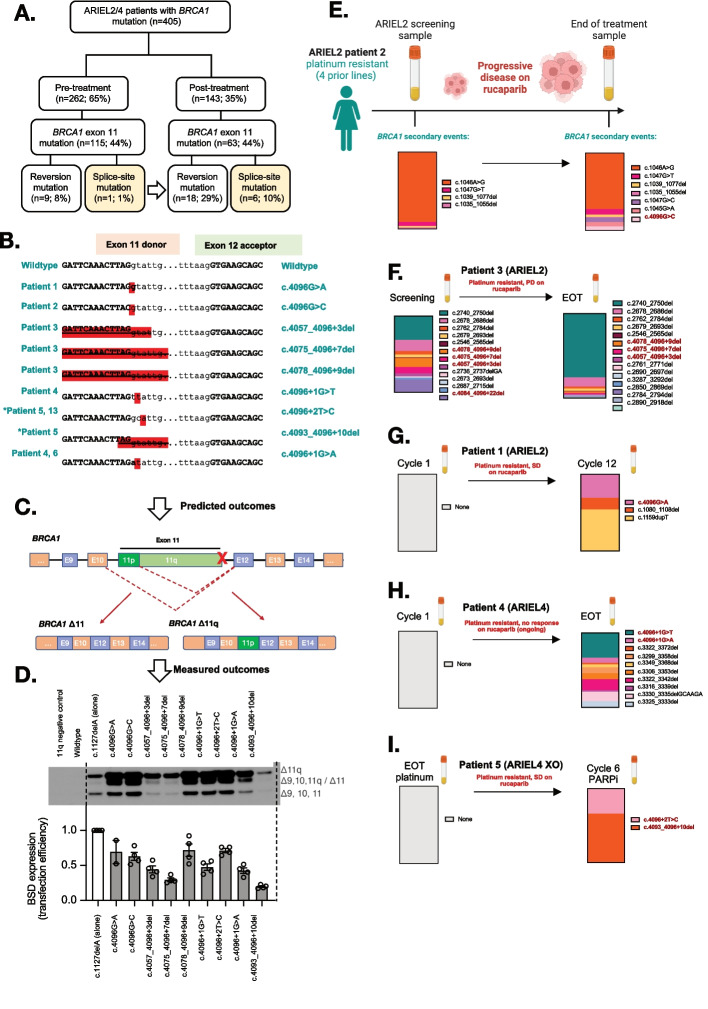
PARP inhibitor (PARPi) therapy has transformed outcomes for patients with homologous recombination DNA repair (HRR) deficient ovarian cancers, for example those with BRCA1 or BRCA2 gene defects. Unfortunately, PARPi resistance is common. Multiple resistance mechanisms have been described, including secondary mutations that restore the HR gene reading frame. BRCA1 splice isoforms △11 and △11q can contribute to PARPi resistance by splicing out the mutation-containing exon, producing truncated, partially functional proteins. However, the clinical impacts and underlying drivers of BRCA1 exon skipping are not fully understood.We analyzed nine ovarian and breast cancer patient derived xenografts (PDX) with BRCA1 exon 11 frameshift mutations for exon skipping and therapy response, including a matched PDX pair derived from a patient pre- and post-chemotherapy/PARPi. BRCA1 exon 11 skipping was elevated in PARPi resistant PDX tumors. Two independent PDX models acquired secondary BRCA1 splice site mutations (SSMs) that drive exon skipping, confirmed using qRT-PCR, RNA sequencing, immunoblotting and minigene modelling. CRISPR/Cas9-mediated disruption of splicing functionally validated exon skipping as a mechanism of PARPi resistance. SSMs were also enriched in post-PARPi ovarian cancer patient cohorts from the ARIEL2 and ARIEL4 clinical trials.Few PARPi resistance mechanisms have been confirmed in the clinical setting. While secondary/reversion mutations typically restore a gene's reading frame, we have identified secondary mutations in patient cohorts that hijack splice sites to enhance mutation-containing exon skipping, resulting in the overexpression of BRCA1 hypomorphs, which in turn promote PARPi resistance. Thus, BRCA1 SSMs can and should be clinically monitored, along with frame-restoring secondary mutations.
© 2024. The Author(s).
Conflict of interest statement
K. Nesic reports nonfinancial support from Clovis Oncology during the conduct of the study. O. Kondrashova reports personal fees from XING Technologies outside the submitted work. K. Nesic, C. Vandenberg, M.J. Wakefield, C.L. Scott, K. Shield-Artin, A. Farrell, E. Kyran and H.E. Barker all receive research support outside of this study from Eisai Inc, AstraZeneca, Boehringer Ingelheim and Ideaya Biosciences. N. Traficante reports grants from AstraZeneca Pty Ltd. during the conduct of the study and grants from AstraZeneca Pty Ltd. outside the submitted work. D. Bowtell reports grants from AstraZeneca Pty Ltd. during the conduct of the study and research support grants from AstraZeneca, Roche-Genentech and BeiGene (paid to institution) outside the submitted work; and personal consulting fees from Exo Therapeutics, that are outside the submitted work. Australian Ovarian Cancer Study reports grants from AstraZeneca Pty Ltd. during the conduct of the study and grants from AstraZeneca Pty Ltd. outside the submitted work. There are no other conflicts of interest in relation to the work under consideration for publication, nor other relationships / conditions / circumstances that present a potential conflict of interest. A. DeFazio reports grants from AstraZeneca outside the submitted work. A. Dobrovic reports grants from National Breast Cancer Foundation of Australia during the conduct of the study. T.C. Harding, K. Lin and T. Kwan were employees of Clovis Oncology. R. Kristeleit reports funding from Clovis Oncology, AstraZeneca, GSK, Pharma&, and was a Co-ordinating Investigator for the ARIEL4 and ATHENA trials. I. McNeish reports advisory boards for Clovis Oncology, AstraZeneca, GSK, OncoC4, Theolytics, Epsila Bio, Duke St Bio, Scancell, Roche, Takeda, and also institutional grant income from AstraZeneca. E.M. Swisher is on the Scientific Advisory Board for Ideaya Biosciences. C.L. Scott reports research support from AstraZeneca Pty Ltd, Boehringer Ingelheim and Eisai Inc, and other support from Clovis Oncology and Sierra Oncology outside the submitted work; and unpaid advisory boards: AstraZeneca, Clovis Oncology, Roche, Eisai, Sierra Oncology, Takeda, MSD. No disclosures were reported by the other authors.
Figures
Update of
-
BRCA1 secondary splice-site mutations drive exon-skipping and PARP inhibitor resistance.medRxiv [Preprint]. 2023 Aug 28:2023.03.20.23287465. doi: 10.1101/2023.03.20.23287465. medRxiv. 2023. Update in: Mol Cancer. 2024 Aug 5;23(1):158. doi: 10.1186/s12943-024-02048-1. PMID: 36993400 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
