

Acid sphingomyelinase deficiency in France: a retrospective survival study
- PMID: 39103853
- PMCID: PMC11301966
- DOI: 10.1186/s13023-024-03234-6
Acid sphingomyelinase deficiency in France: a retrospective survival study
Abstract
Background: Acid sphingomyelinase deficiency (ASMD) or Niemann-Pick disease types A, A/B, and B is a progressive, life-limiting, autosomal recessive disorder caused by sphingomyelin phosphodiesterase 1 (SMPD1) gene mutations. There is a need to increase the understanding of morbidity and mortality across children to adults diagnosed with ASMD.
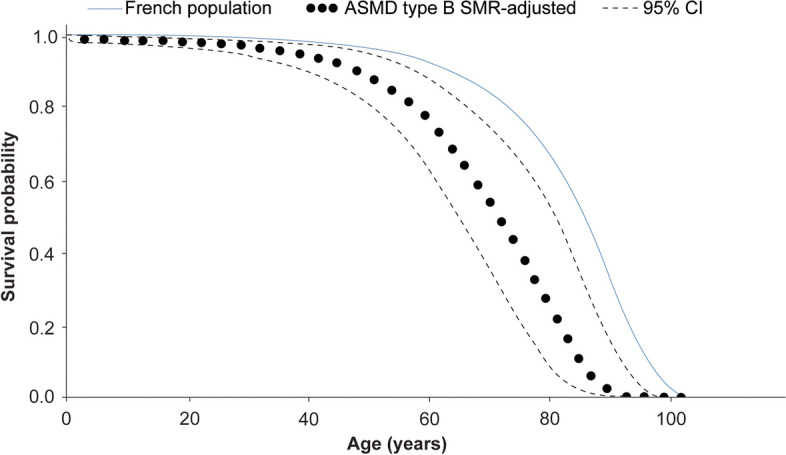
Methods: This observational retrospective survey analysed medical records of patients with ASMD with retrievable data from 27 hospitals in France, diagnosed/followed up between 1st January 1990 and 31st December 2020. Eligible records were abstracted to collect demographic, medical/developmental history, and mortality data. Survival outcomes were estimated from birth until death using Kaplan-Meier survival analyses; standardised mortality ratio (SMR) was also explored.
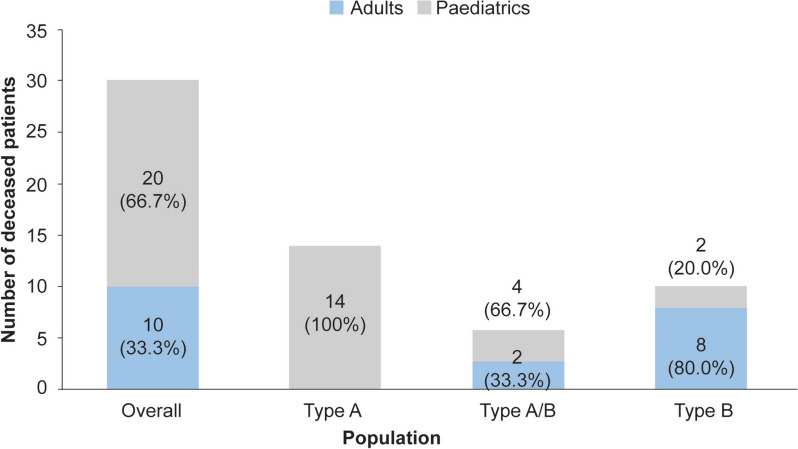
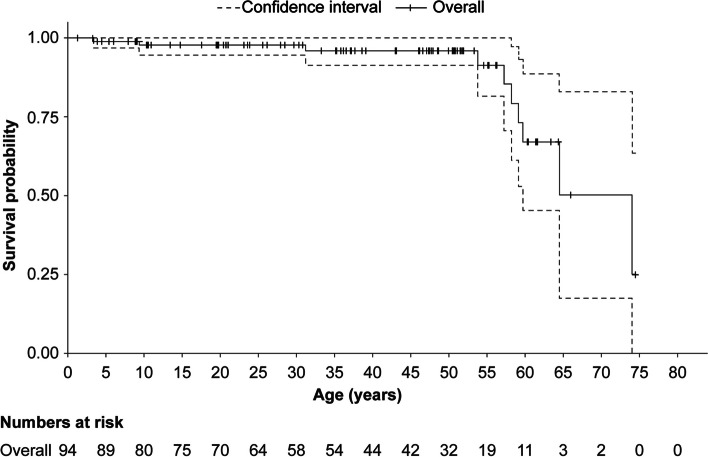
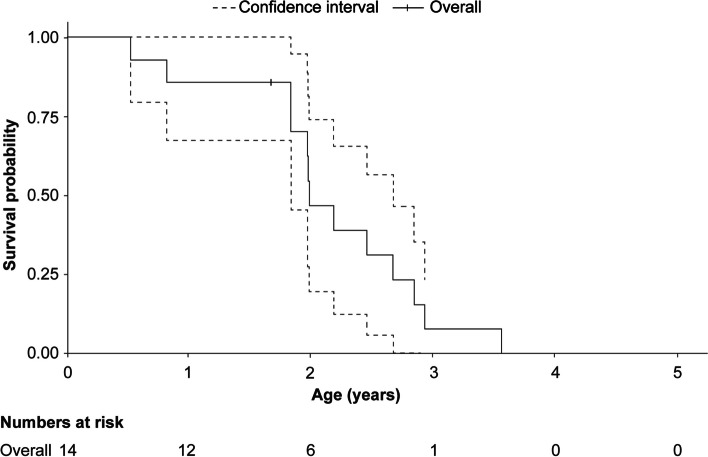
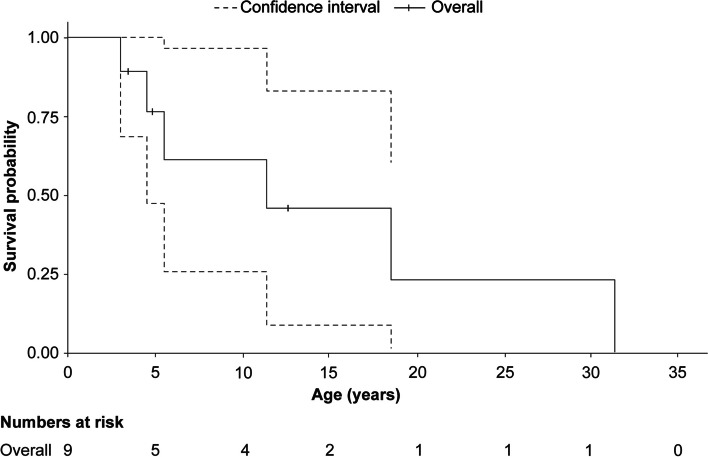
Results: A total of 118 medical records of patients with ASMD (type B [n = 94], type A [n = 15], and type A/B [n = 9]) were assessed. The majority of patients were males (63.6%); the median [range] age at diagnosis was 8.0 [1.0-18.0] months (type A), 1.0 [0-3] year (type A/B), and 5.5 [0-73] years (type B). Overall, 30 patients were deceased at the study completion date; the median [range] age at death for patients with ASMD type A (n = 14) was 1 [0-3.6] year, type A/B (n = 6) was 8.5 [3.0-30.9] years, and type B (n = 10) was 57.6 [3.4-74.1] years. The median [95% confidence interval (CI)] survival age from birth in patients with ASMD type A and type A/B was 2.0 [1.8-2.7] years and 11.4 [5.5-18.5] years, respectively. Survival analysis in ASMD type B was explored using SMR [95% CI] analysis (3.5 [1.6-5.9]), which showed that age-specific deaths in the ASMD type B population were 3.5 times more frequent than those in the general French population. The causes of death were mostly severe progressive neurodegeneration (type A: 16.7%), cancer (type B: 16.7%), or unspecified (across groups: 33.3%).
Conclusions: This study illustrated a substantial burden of illness with high mortality rates in patients with ASMD, including adults with ASMD type B, in France.
Keywords: Acid sphingomyelinase deficiency (ASMD); France; Mortality; Niemann–Pick disease; Niemann–Pick disease type B; Standardised mortality ratio; Survival.
© 2024. The Author(s).
Conflict of interest statement
WM has received honoraria from Amicus, Sanofi, Chiesi and Takeda; consultation fees from Chiesi and Sanofi. NG has received honoraria or consultation fees from Sanofi, Ultragenyx, and Chiesi; PI in ASCEND-PED, a Sanofi-sponsored trial. MTV has received honoraria from Orphazyme, Orchard Therapeutics, and Sanofi. RF has received honoraria from Sanofi, Takeda, and Chiesi; travel fees and accommodation from Amicus. AC has received honoraria from Sanofi. CD has received honoraria from Ultragenyx and Sanofi. CL has received honoraria or consultation fees from Sanofi and Boehringer Ingelheim. BH has received honoraria from Takeda and Orchard; travel fees and accommodation from Chiesi, PTC Therapeutics; PI in trials sponsored by JR Pharmaceuticals, Chiesi, Orphazyme, Lysogene and Mallincrodt. NB has received consultation fees for speaking, education, clinical trials and research from Sanofi and Takeda. YU has received consulting fees and honoraria for speaking, or educational events from Boehringer-Ingelheim, Sanofi, GSK, Pfizer, and Oxyvie. DL has participated in a Board with Sanofi. TL has received consulting fees from Sanofi and Chiesi. AD and RP-J are employees and may hold stocks and/or stock options in Sanofi. FL was an employee of Sanofi at the time of study conduct. SP has received honoraria from Sanofi and Biomarin; travel fees and accommodation from Sanofi, Biomarin and Takeda. OL has received honoraria from Amicus, Sanofi, and Chiesi; PI in ASCEND, a Sanofi-sponsored trial; consultation fees from Amicus and Sanofi.
Figures
References
-
- Schuchman EH, Desnick RJ. Niemann-Pick disease types A and B: Acid sphingomyelinase deficiencies. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA, editors. The online metabolic and molecular bases of inherited disease. New York, NY: McGraw-Hill Education; 2019.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
