Myasthenia gravis: the future is here
- PMID: 39105625
- PMCID: PMC11178544
- DOI: 10.1172/JCI179742
Myasthenia gravis: the future is here
Abstract
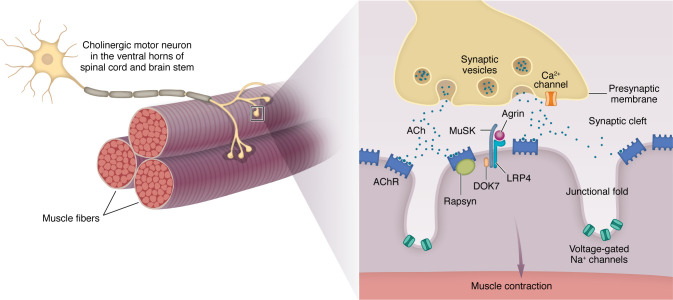
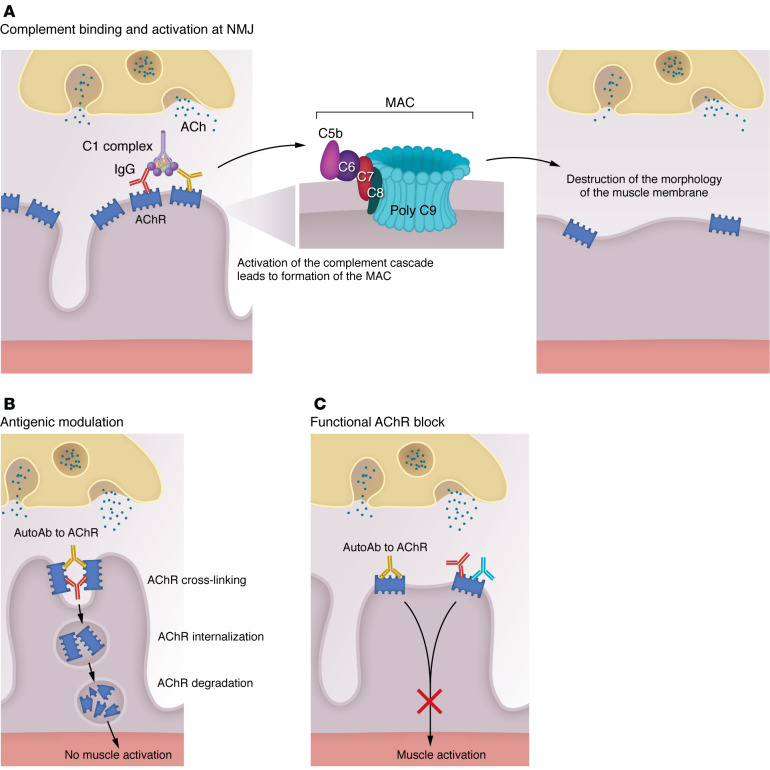
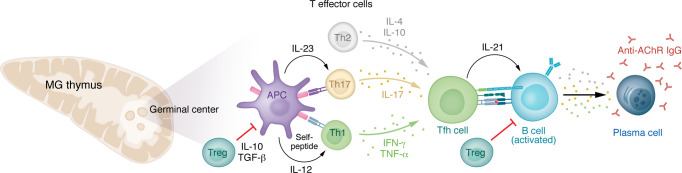
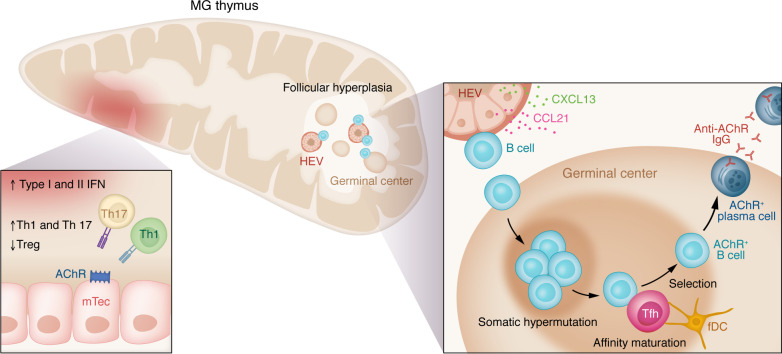
Myasthenia gravis (MG) stands as a prototypical antibody-mediated autoimmune disease: it is dependent on T cells and characterized by the presence of autoantibodies targeting proteins located on the postsynaptic surface of skeletal muscle, known as the neuromuscular junction. Patients with MG exhibit a spectrum of weakness, ranging from limited ocular muscle involvement to life-threatening respiratory failure. Recent decades have witnessed substantial progress in understanding the underlying pathophysiology, leading to the delineation of distinct subcategories within MG, including MG linked to AChR or MuSK antibodies as well as age-based distinction, thymoma-associated, and immune checkpoint inhibitor-induced MG. This heightened understanding has paved the way for the development of more precise and targeted therapeutic interventions. Notably, the FDA has recently approved therapeutic inhibitors of complement and the IgG receptor FcRn, a testament to our improved comprehension of autoantibody effector mechanisms in MG. In this Review, we delve into the various subgroups of MG, stratified by age, autoantibody type, and histology of the thymus with neoplasms. Furthermore, we explore both current and potential emerging therapeutic strategies, shedding light on the evolving landscape of MG treatment.
Conflict of interest statement
Figures