

Diagnostic utility of DNA methylation analysis in genetically unsolved pediatric epilepsies and CHD2 episignature refinement
- PMID: 39107278
- PMCID: PMC11303402
- DOI: 10.1038/s41467-024-50159-6
Diagnostic utility of DNA methylation analysis in genetically unsolved pediatric epilepsies and CHD2 episignature refinement
Abstract
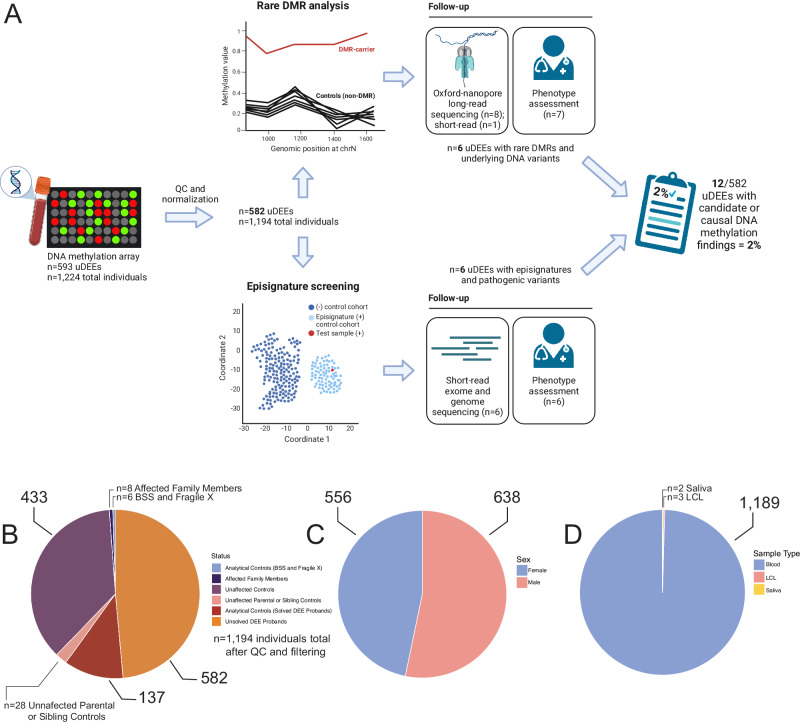
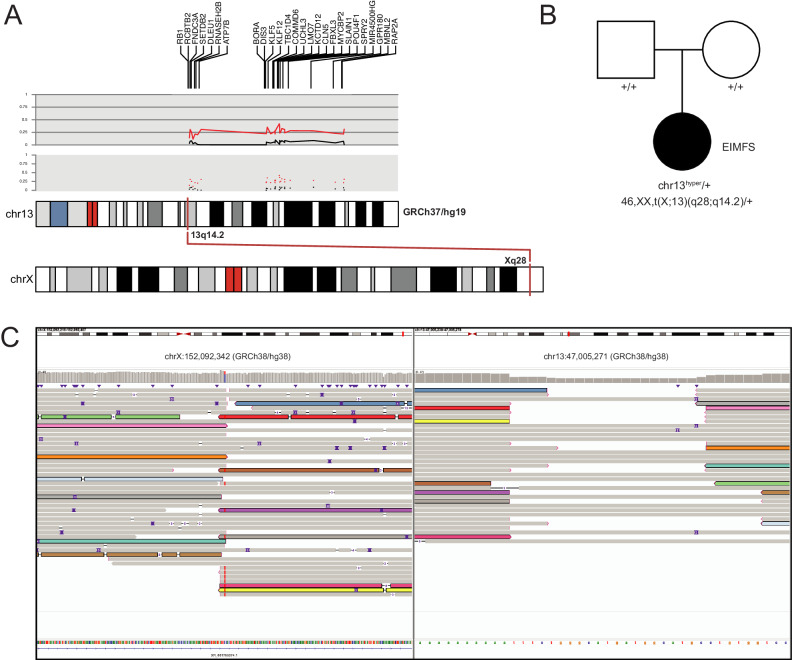
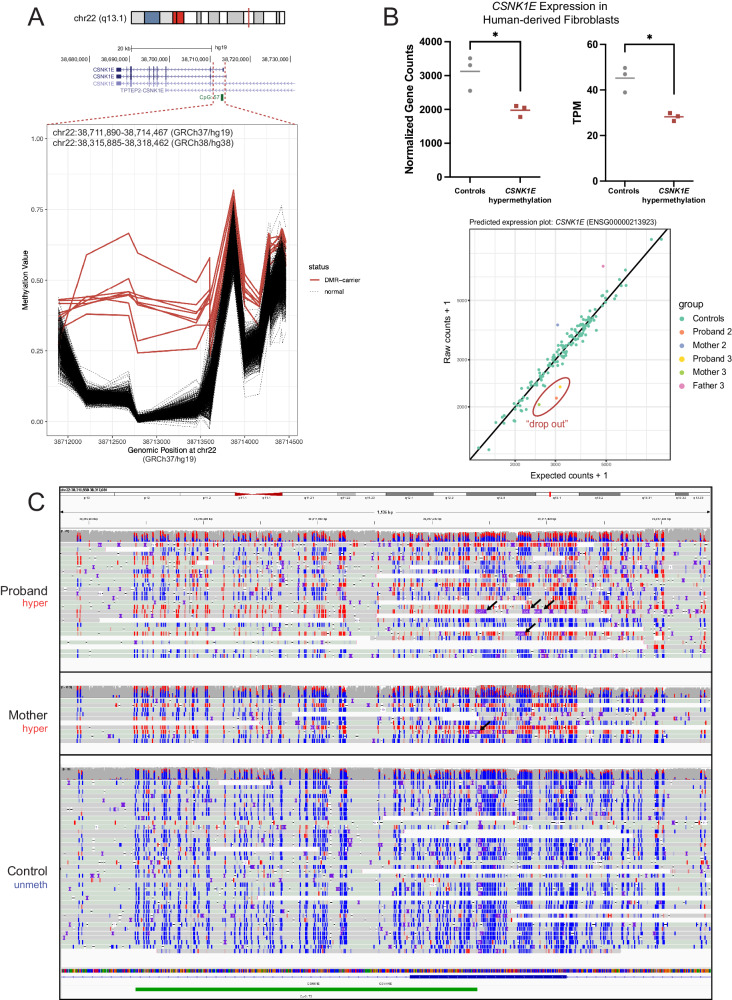
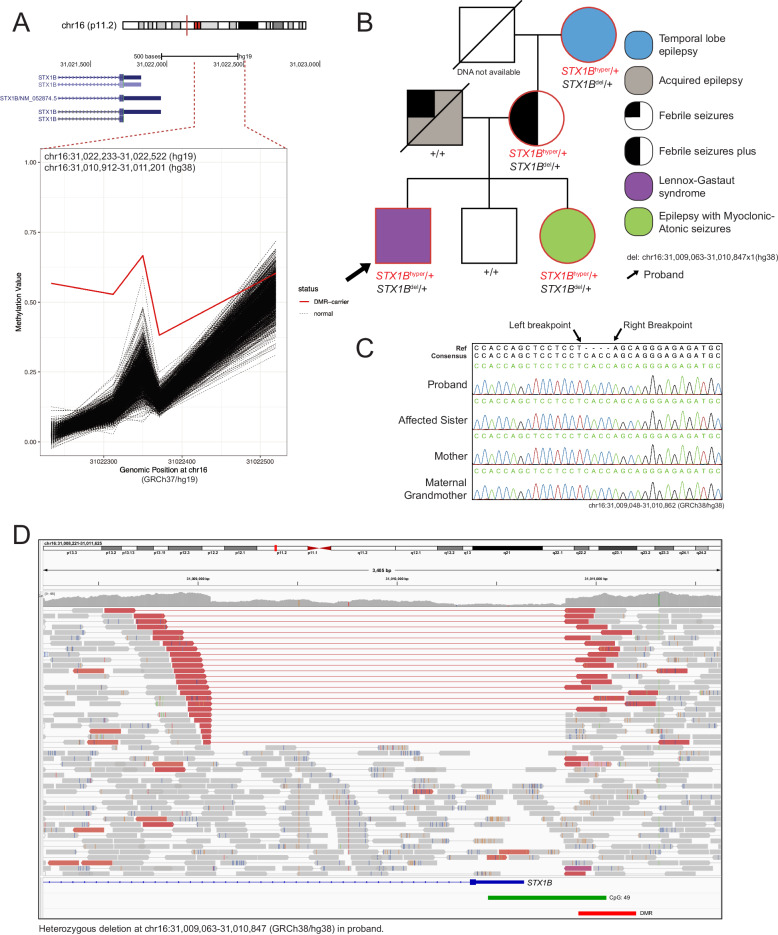
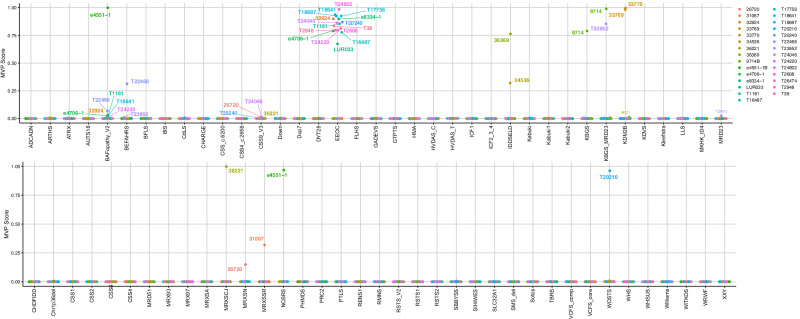
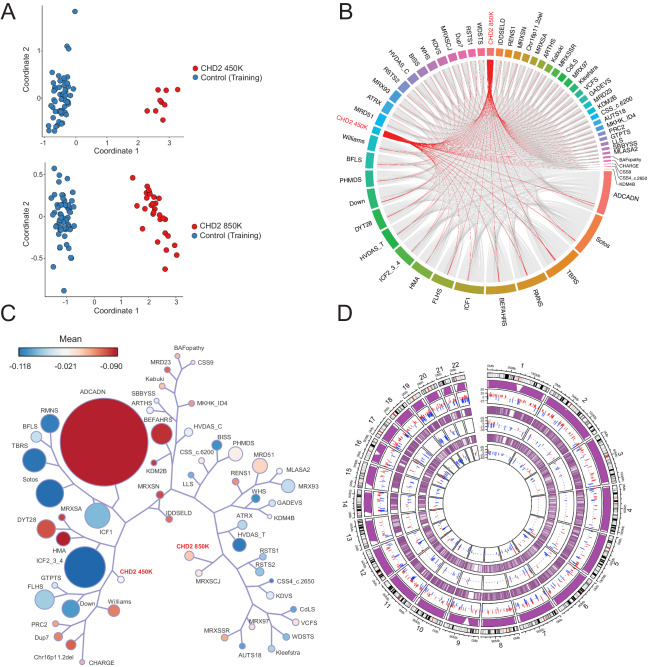
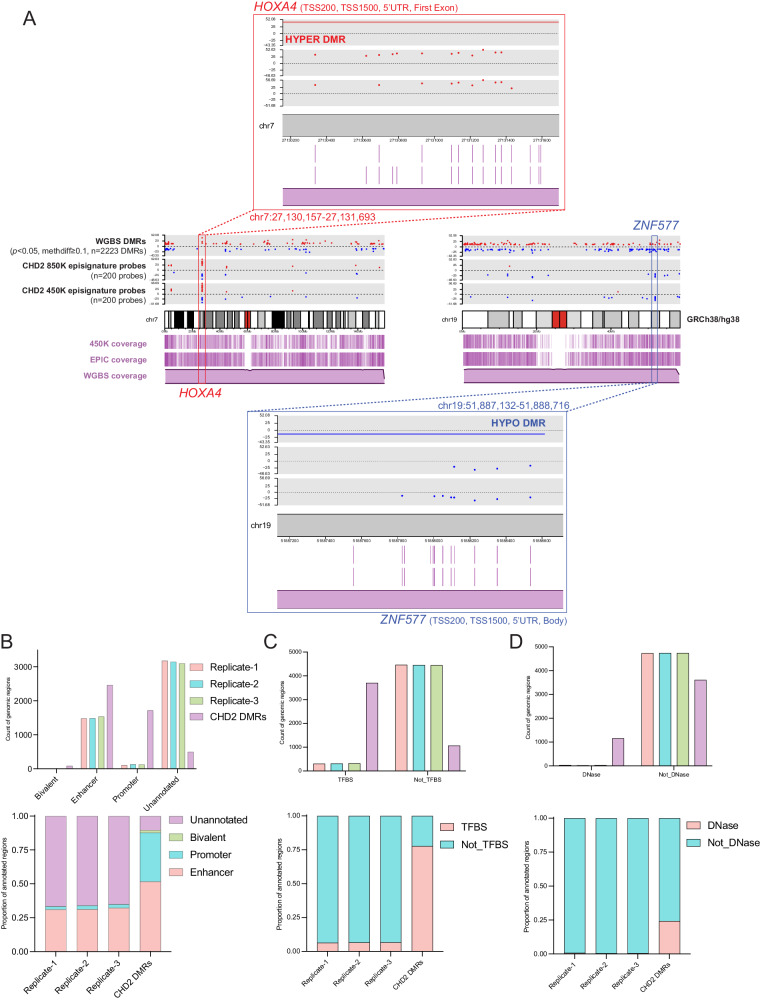
Sequence-based genetic testing identifies causative variants in ~ 50% of individuals with developmental and epileptic encephalopathies (DEEs). Aberrant changes in DNA methylation are implicated in various neurodevelopmental disorders but remain unstudied in DEEs. We interrogate the diagnostic utility of genome-wide DNA methylation array analysis on peripheral blood samples from 582 individuals with genetically unsolved DEEs. We identify rare differentially methylated regions (DMRs) and explanatory episignatures to uncover causative and candidate genetic etiologies in 12 individuals. Using long-read sequencing, we identify DNA variants underlying rare DMRs, including one balanced translocation, three CG-rich repeat expansions, and four copy number variants. We also identify pathogenic variants associated with episignatures. Finally, we refine the CHD2 episignature using an 850 K methylation array and bisulfite sequencing to investigate potential insights into CHD2 pathophysiology. Our study demonstrates the diagnostic yield of genome-wide DNA methylation analysis to identify causal and candidate variants as 2% (12/582) for unsolved DEE cases.
© 2024. The Author(s).
Conflict of interest statement
B.S. is a shareholder in EpiSign Inc, a company involved in commercialization of EpiSignTM software. D.E.M. is on a scientific advisory board at ONT and has received travel support from ONT to speak on their behalf. D.E.M. is engaged in a research agreement with ONT. D.E.M. holds stock options in MyOme. I.E.S. has served on scientific advisory boards for BioMarin, Chiesi, Eisai, Encoded Therapeutics, GlaxoSmithKline, Knopp Biosciences, Nutricia, Rogcon, Takeda Pharmaceuticals, UCB, Xenon Pharmaceuticals, Cerecin; has received speaker honoraria from GlaxoSmithKline, UCB, BioMarin, Biocodex, Chiesi, Liva Nova, Nutricia, Zuellig Pharma, Stoke Therapeutics and Eisai; has received funding for travel from UCB, Biocodex, GlaxoSmithKline, Biomarin, Encoded Therapeutics Stoke Therapeutics and Eisai; has served as an investigator for Anavex Life Sciences, Cerevel Therapeutics, Eisai, Encoded Therapeutics, EpiMinder Inc, Epygenyx, ES-Therapeutics, GW Pharma, Marinus, Neurocrine BioSciences, Ovid Therapeutics, Takeda Pharmaceuticals, UCB, Ultragenyx, Xenon Pharmaceuticals, Zogenix and Zynerba; has consulted for Care Beyond Diagnosis, Epilepsy Consortium, Atheneum Partners, Ovid Therapeutics, UCB, Zynerba Pharmaceuticals, BioMarin, Encoded Therapeutics and Biohaven Pharmaceuticals; and is a Non-Executive Director of Bellberry Ltd and a Director of the Australian Academy of Health and Medical Sciences and the Australian Council of Learned Academies Limited. I.E.S. may accrue future revenue on pending patent WO61/010176 (filed: 2008): Therapeutic Compound; has a patent for
Figures
Update of
-
Diagnostic Utility of Genome-wide DNA Methylation Analysis in Genetically Unsolved Developmental and Epileptic Encephalopathies and Refinement of a CHD2 Episignature.medRxiv [Preprint]. 2023 Oct 12:2023.10.11.23296741. doi: 10.1101/2023.10.11.23296741. medRxiv. 2023. Update in: Nat Commun. 2024 Aug 6;15(1):6524. doi: 10.1038/s41467-024-50159-6. PMID: 37873138 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
