

This is a preprint.
Maximizing the clinical utility and performance of cytology samples for comprehensive genetic profiling - A report on the impact of process optimization through the analysis of 4,871 cytology samples profiled by MSK-IMPACT
- PMID: 39108489
- PMCID: PMC11302697
- DOI: 10.21203/rs.3.rs-4746484/v1
Maximizing the clinical utility and performance of cytology samples for comprehensive genetic profiling - A report on the impact of process optimization through the analysis of 4,871 cytology samples profiled by MSK-IMPACT
Update in
-
Maximizing the clinical utility and performance of cytology samples for comprehensive genetic profiling.Nat Commun. 2025 Jan 2;16(1):116. doi: 10.1038/s41467-024-55456-8. Nat Commun. 2025. PMID: 39747849 Free PMC article.
Abstract
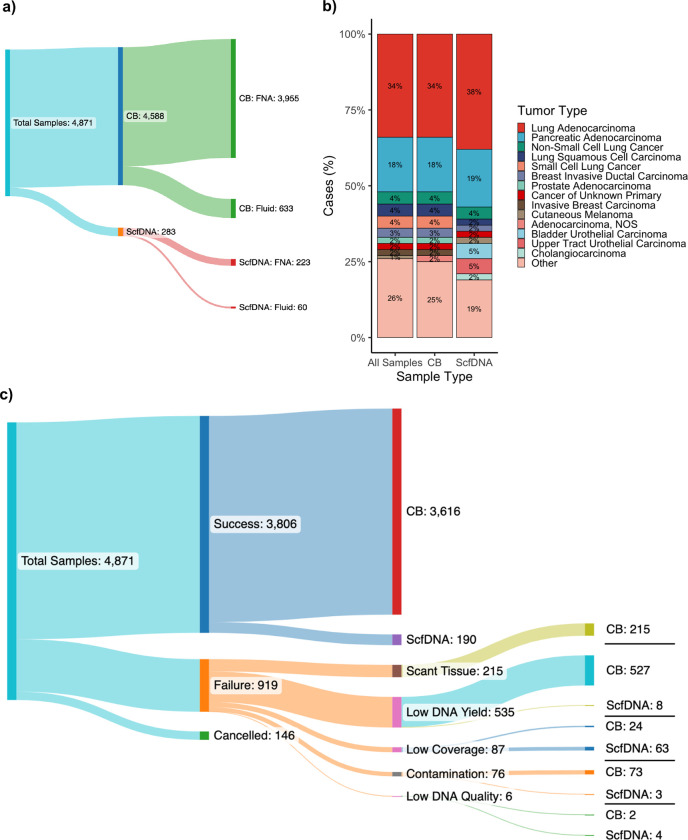
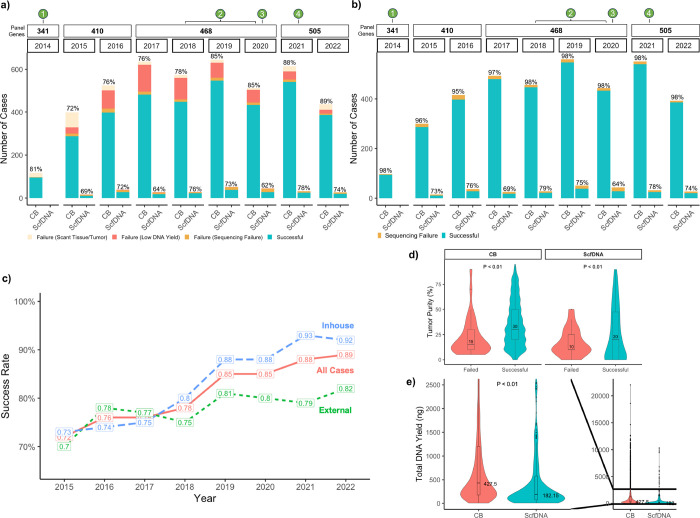
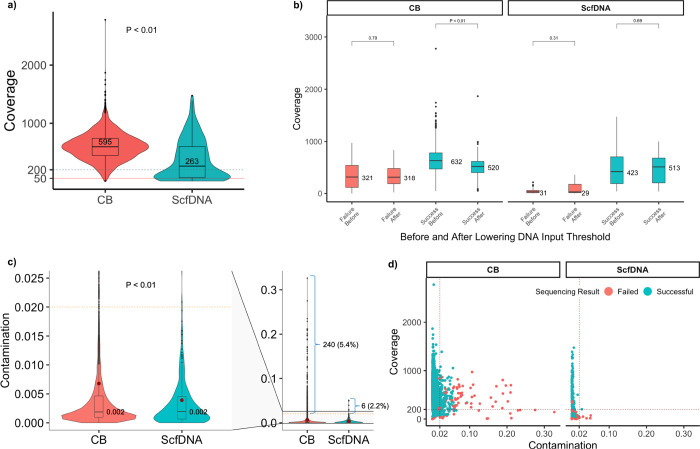
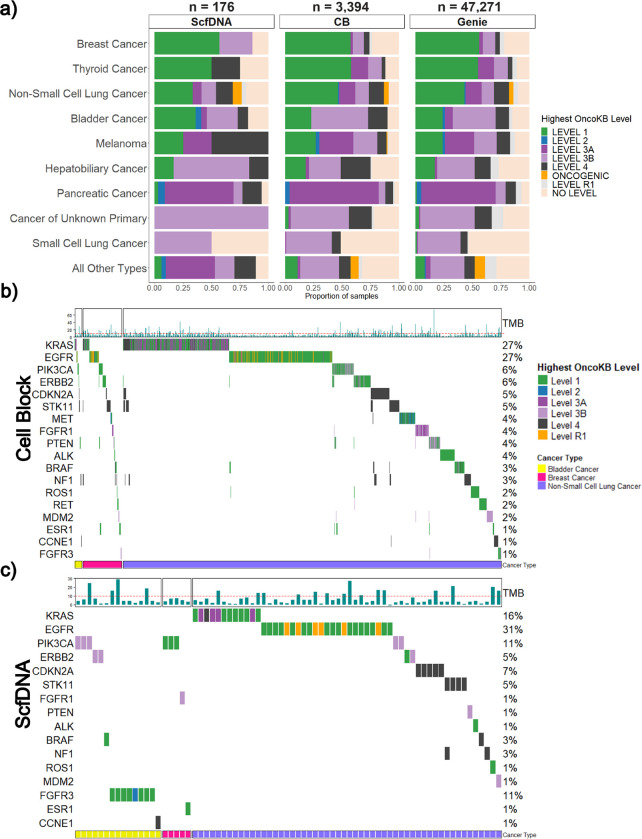
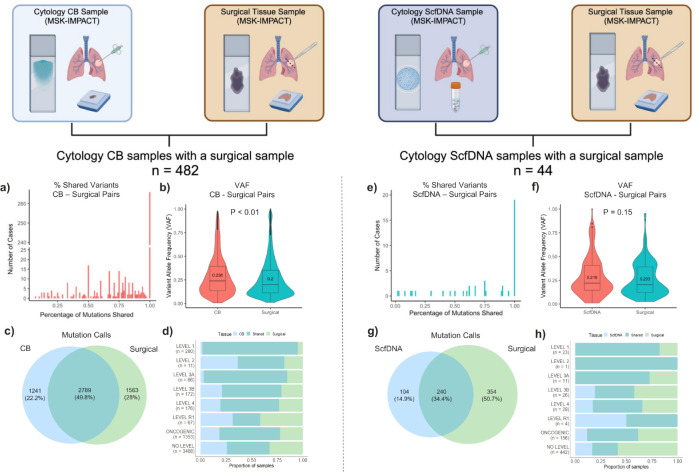
Comprehensive molecular profiling by next generation sequencing (NGS) has revolutionized tumor classification and biomarker evaluation. However, routine implementation is challenged by the scant nature of diagnostic material obtained through minimally invasive procedures. Here, we describe our long-term experience in profiling cytology samples with an in-depth assessment of the performance, quality metrics, biomarker identification capabilities, and potential pitfalls. We highlight the impact of several optimization strategies to maximize performance with 4,871 prospectively sequenced clinical cytology samples tested by MSK-IMPACT™. Special emphasis is given to the use of residual supernatant cell free DNA (ScfDNA) as a valuable source of tumor DNA. Overall, cytology samples were similar in performance to surgical samples in identifying clinically relevant genomic alterations, achieving success rates up to 93% with full optimization. While cell block (CB) samples had excellent performance overall, low-level cross-contamination was identified in a small proportion of cases (4.7%), a common pitfall intrinsic to the processing of paraffin blocks, suggesting that more stringent precautions and processing modifications should be considered in quality control initiatives. By contrast ScfDNA samples had negligible contamination. Finally, ScfDNA testing exclusively used as a rescue strategy delivered successful results in 71% of cases where tumor tissue from CB was depleted.
Keywords: Cell-free DNA; Cytology; Molecular Cytology; Next-Generation Sequencing; Supernatant Fluid.
Figures
References
-
- Garraway LA (2013) Genomics-driven oncology: framework for an emerging paradigm. J Clin Oncol 31:1806–1814 - PubMed
-
- Wakai T et al. (2019) Next-generation sequencing-based clinical sequencing: toward precision medicine in solid tumors. Int J Clin Oncol 24:115–122 - PubMed
-
- Baum JE et al. (2017) Accuracy of next-generation sequencing for the identification of clinically relevant variants in cytology smears in lung adenocarcinoma. Cancer Cytopathol 125:398–406 - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
