

Assessing predictions on fitness effects of missense variants in HMBS in CAGI6
- PMID: 39110250
- PMCID: PMC12085147
- DOI: 10.1007/s00439-024-02680-3
Assessing predictions on fitness effects of missense variants in HMBS in CAGI6
Abstract
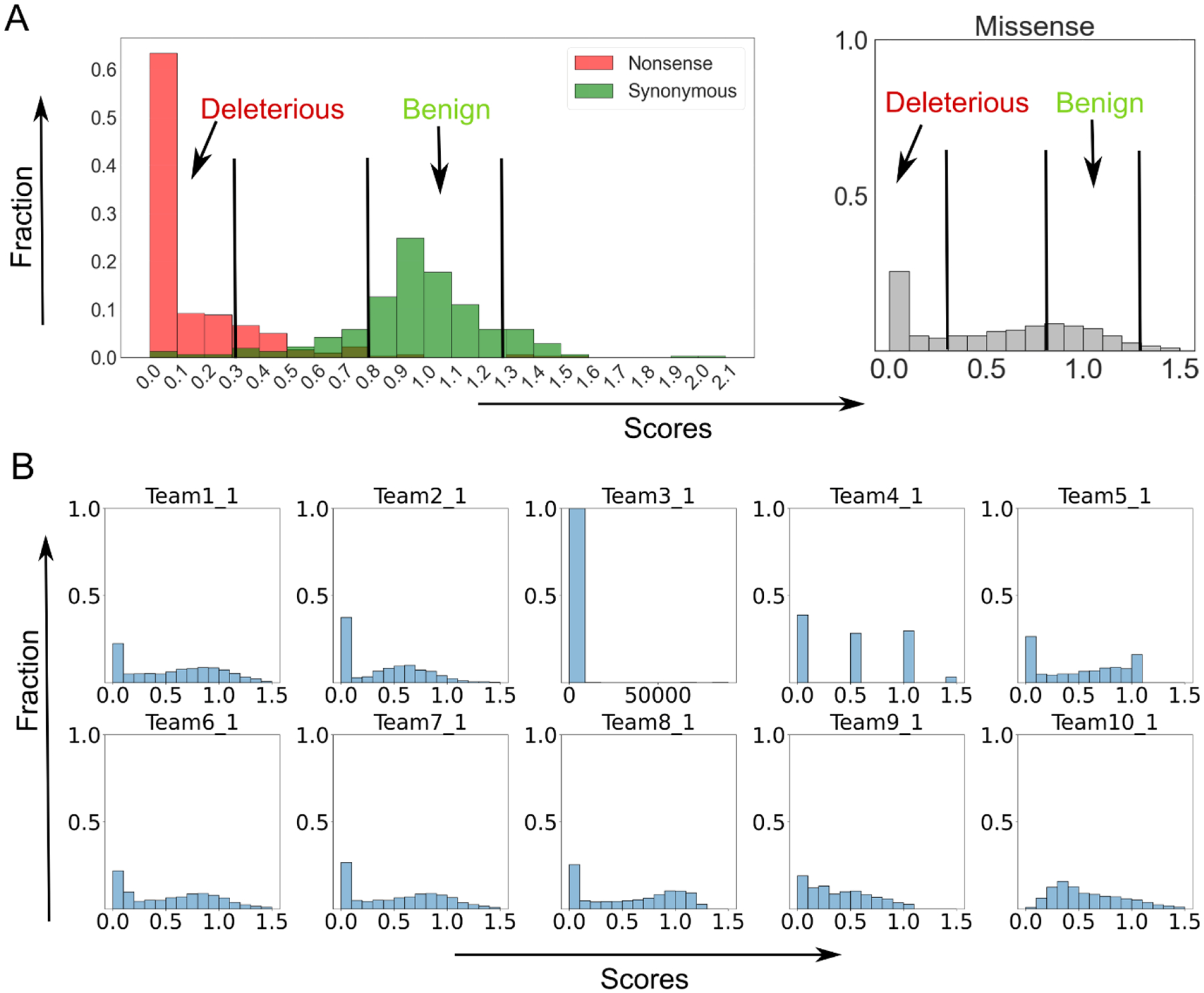
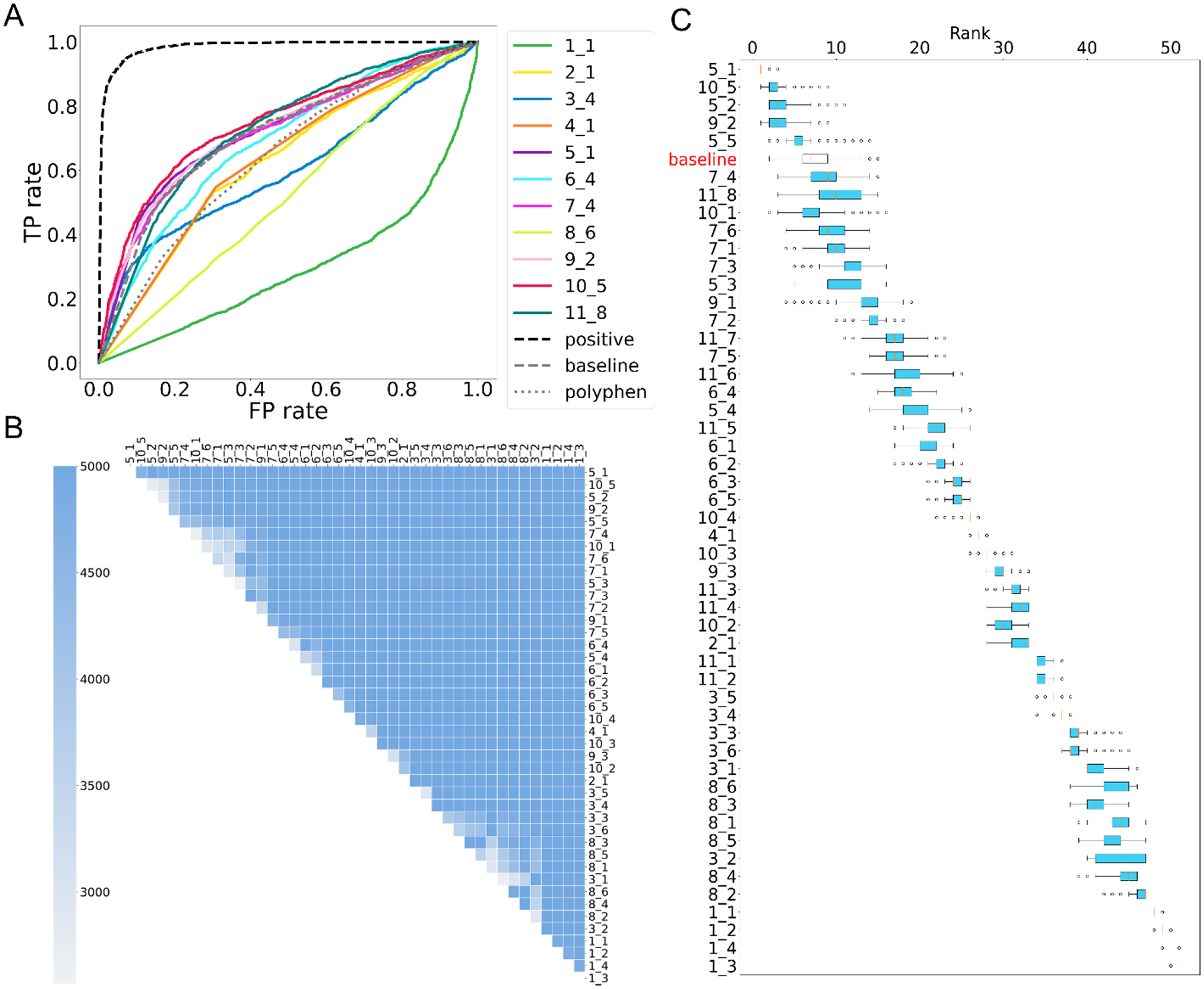
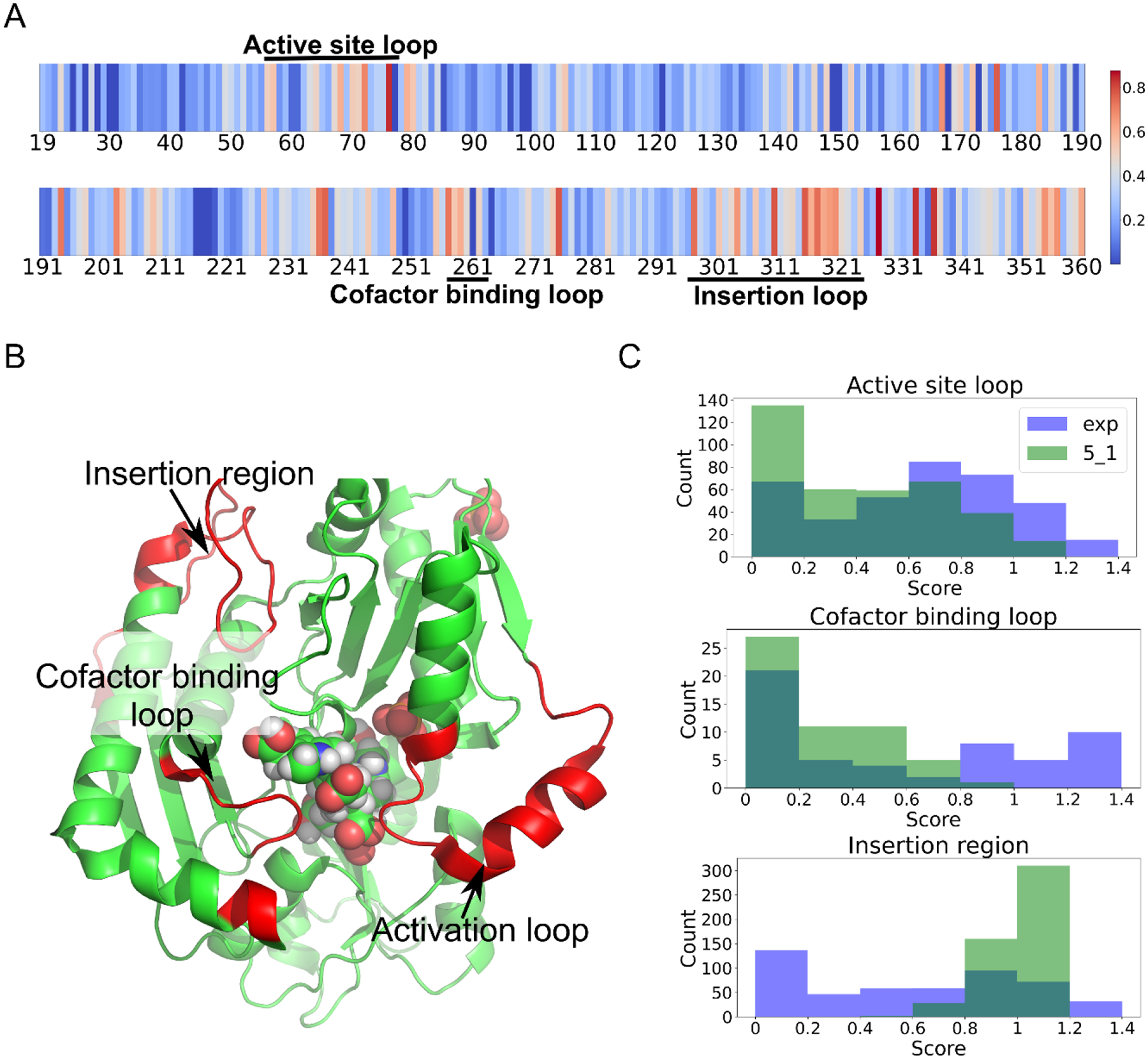
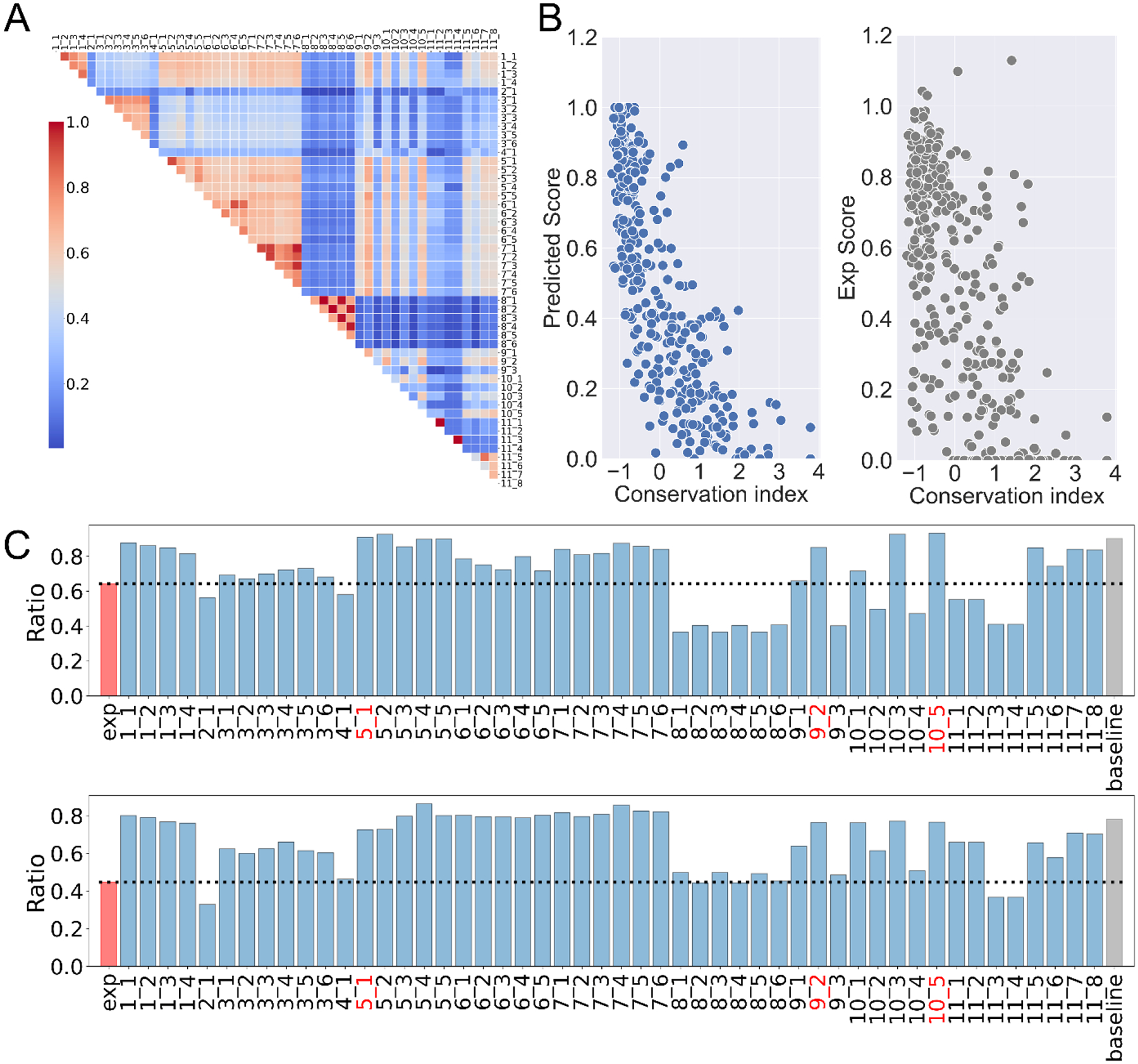
This paper presents an evaluation of predictions submitted for the "HMBS" challenge, a component of the sixth round of the Critical Assessment of Genome Interpretation held in 2021. The challenge required participants to predict the effects of missense variants of the human HMBS gene on yeast growth. The HMBS enzyme, critical for the biosynthesis of heme in eukaryotic cells, is highly conserved among eukaryotes. Despite the application of a variety of algorithms and methods, the performance of predictors was relatively similar, with Kendall's tau correlation coefficients between predictions and experimental scores around 0.3 for a majority of submissions. Notably, the median correlation (≥ 0.34) observed among these predictors, especially the top predictions from different groups, was greater than the correlation observed between their predictions and the actual experimental results. Most predictors were moderately successful in distinguishing between deleterious and benign variants, as evidenced by an area under the receiver operating characteristic (ROC) curve (AUC) of approximately 0.7 respectively. Compared with the recent two rounds of CAGI competitions, we noticed more predictors outperformed the baseline predictor, which is solely based on the amino acid frequencies. Nevertheless, the overall accuracy of predictions is still far short of positive control, which is derived from experimental scores, indicating the necessity for considerable improvements in the field. The most inaccurately predicted variants in this round were associated with the insertion loop, which is absent in many orthologs, suggesting the predictors still heavily rely on the information from multiple sequence alignment.
© 2024. The Author(s), under exclusive licence to Springer-Verlag GmbH Germany, part of Springer Nature.
Conflict of interest statement
Declarations. Conflcit of interest: The authors have not disclosed any competing interests.
Figures
Similar articles
-
FiTMuSiC: leveraging structural and (co)evolutionary data for protein fitness prediction.Hum Genomics. 2024 Apr 16;18(1):36. doi: 10.1186/s40246-024-00605-9. Hum Genomics. 2024. PMID: 38627807 Free PMC article.
-
Assessing predictions on fitness effects of missense variants in calmodulin.Hum Mutat. 2019 Sep;40(9):1463-1473. doi: 10.1002/humu.23857. Epub 2019 Sep 3. Hum Mutat. 2019. PMID: 31283071 Free PMC article.
-
Assessing predictions of fitness effects of missense mutations in SUMO-conjugating enzyme UBE2I.Hum Mutat. 2017 Sep;38(9):1051-1063. doi: 10.1002/humu.23293. Hum Mutat. 2017. PMID: 28817247 Free PMC article.
-
Objective assessment of the evolutionary action equation for the fitness effect of missense mutations across CAGI-blinded contests.Hum Mutat. 2017 Sep;38(9):1072-1084. doi: 10.1002/humu.23266. Epub 2017 Jun 21. Hum Mutat. 2017. PMID: 28544059 Free PMC article. Review.
-
Analysis and Interpretation of the Impact of Missense Variants in Cancer.Int J Mol Sci. 2021 May 21;22(11):5416. doi: 10.3390/ijms22115416. Int J Mol Sci. 2021. PMID: 34063805 Free PMC article. Review.
Cited by
-
FiTMuSiC: leveraging structural and (co)evolutionary data for protein fitness prediction.Hum Genomics. 2024 Apr 16;18(1):36. doi: 10.1186/s40246-024-00605-9. Hum Genomics. 2024. PMID: 38627807 Free PMC article.
References
-
- Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, Kanavy DM, Luo X, McNulty SM, Starita LM, Tavtigian SV, Wright MW, Harrison SM, Biesecker LG, Berg JS, Clinical Genome Resource Sequence Variant Interpretation Working G (2019) Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med 12: 3. doi: 10.1186/s13073-019-0690-2 - DOI - PMC - PubMed
MeSH terms
Grants and funding
- U24 HG007346/NH/NIH HHS/United States
- GM127390/NH/NIH HHS/United States
- U01 HG012022/HG/NHGRI NIH HHS/United States
- R35 GM127390/GM/NIGMS NIH HHS/United States
- R35 GM134922/GM/NIGMS NIH HHS/United States
- I-1505/Welch Foundation
- HG012022/NH/NIH HHS/United States
- 2224128/National Science Foundation
- R35GM124952/NH/NIH HHS/United States
- R35-GM134922/NH/NIH HHS/United States
- MIUR-PRIN-201744NR8S/Ministero dell'Istruzione e del Merito
- RP210041/Cancer Prevention and Research Institute of Texas
- U24 HG007346/HG/NHGRI NIH HHS/United States
- R35 GM124952/GM/NIGMS NIH HHS/United States
- I-2095-20220331/Welch Foundation
LinkOut - more resources
Full Text Sources
