

SYNGAP1 deficiency disrupts synaptic neoteny in xenotransplanted human cortical neurons in vivo
- PMID: 39111306
- PMCID: PMC11446607
- DOI: 10.1016/j.neuron.2024.07.007
SYNGAP1 deficiency disrupts synaptic neoteny in xenotransplanted human cortical neurons in vivo
Abstract
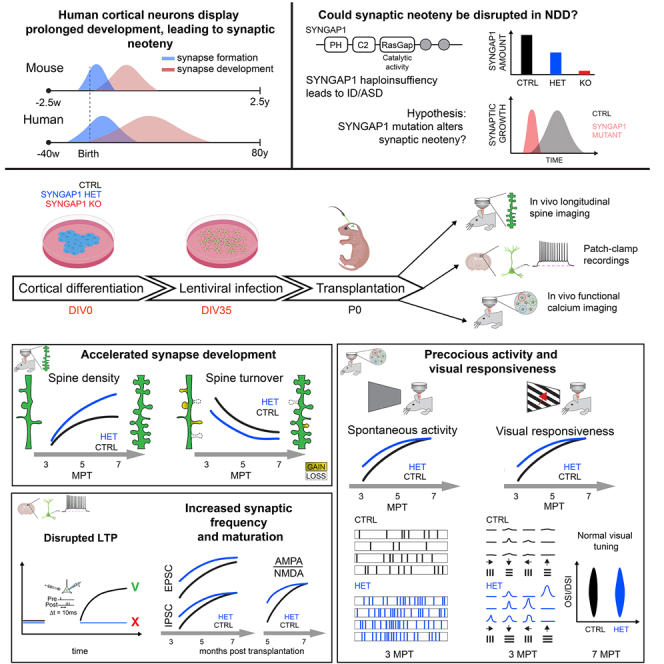
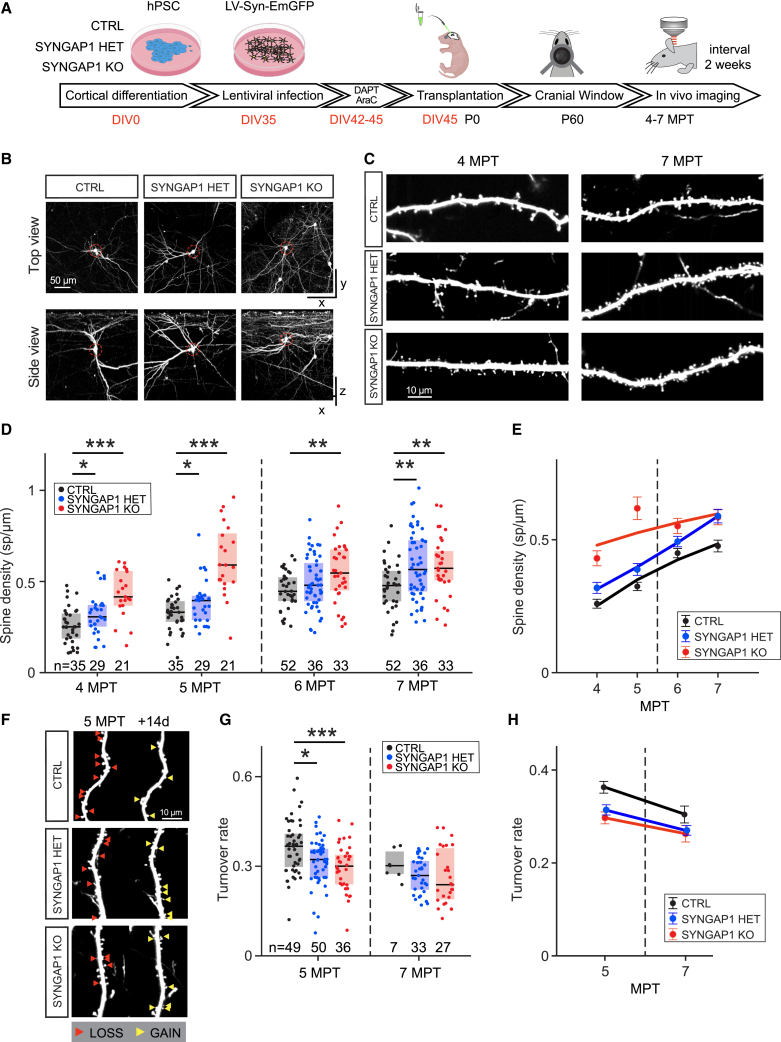
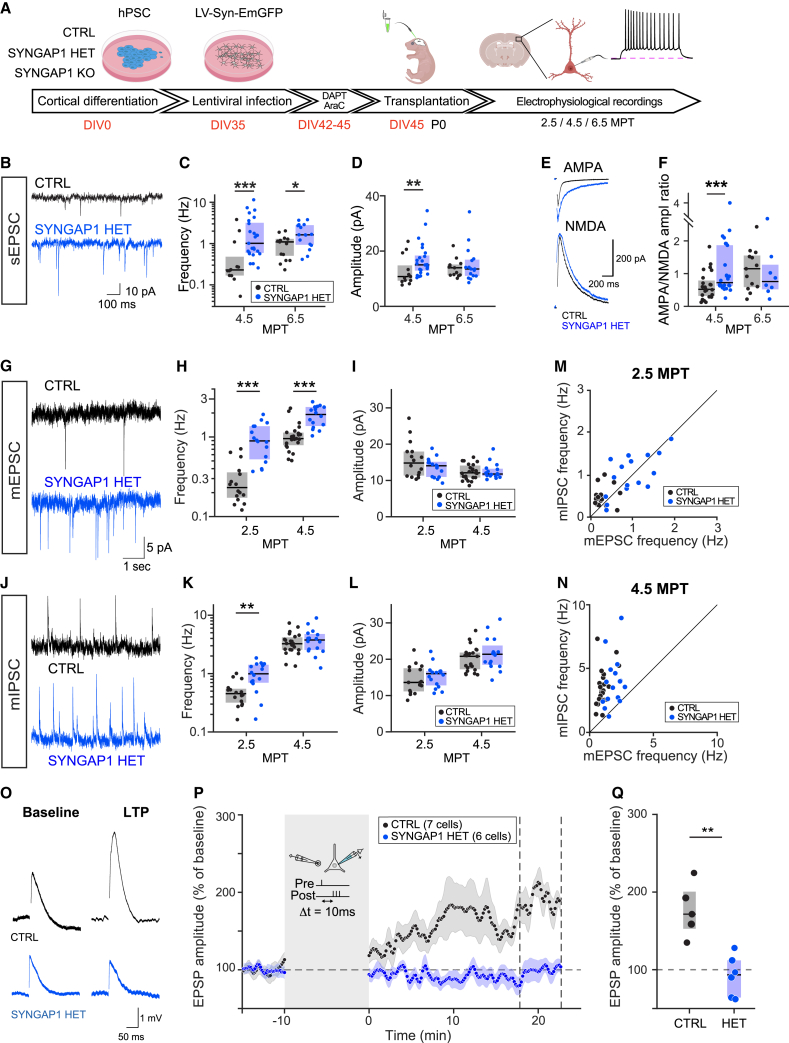
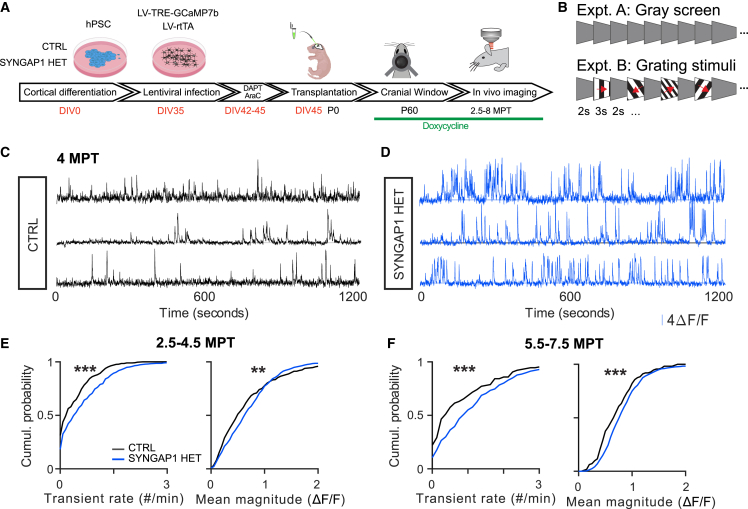
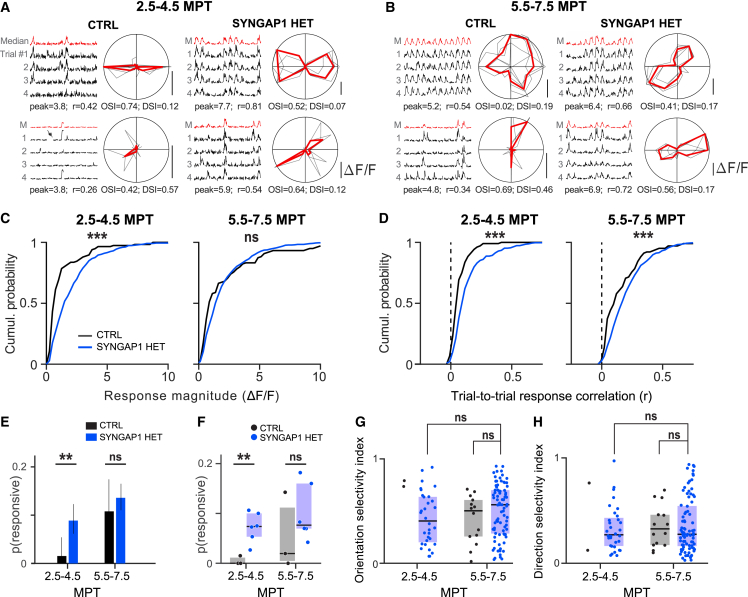
Human brain ontogeny is characterized by a considerably prolonged neotenic development of cortical neurons and circuits. Neoteny is thought to be essential for the acquisition of advanced cognitive functions, which are typically altered in intellectual disability (ID) and autism spectrum disorders (ASDs). Human neuronal neoteny could be disrupted in some forms of ID and/or ASDs, but this has never been tested. Here, we use xenotransplantation of human cortical neurons into the mouse brain to model SYNGAP1 haploinsufficiency, one of the most prevalent genetic causes of ID/ASDs. We find that SYNGAP1-deficient human neurons display strong acceleration of morphological and functional synaptic formation and maturation alongside disrupted synaptic plasticity. At the circuit level, SYNGAP1-haploinsufficient neurons display precocious acquisition of responsiveness to visual stimulation months ahead of time. Our findings indicate that SYNGAP1 is required cell autonomously for human neuronal neoteny, providing novel links between human-specific developmental mechanisms and ID/ASDs.
Keywords: SYNGAP1; autism spectrum disorder; cerebral cortex; evolution; human brain development; neoteny; neurodevelopmental disease.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no conflicts of interest.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
