

Molecular mimicry in multisystem inflammatory syndrome in children
- PMID: 39112696
- PMCID: PMC11324515
- DOI: 10.1038/s41586-024-07722-4
Molecular mimicry in multisystem inflammatory syndrome in children
Abstract
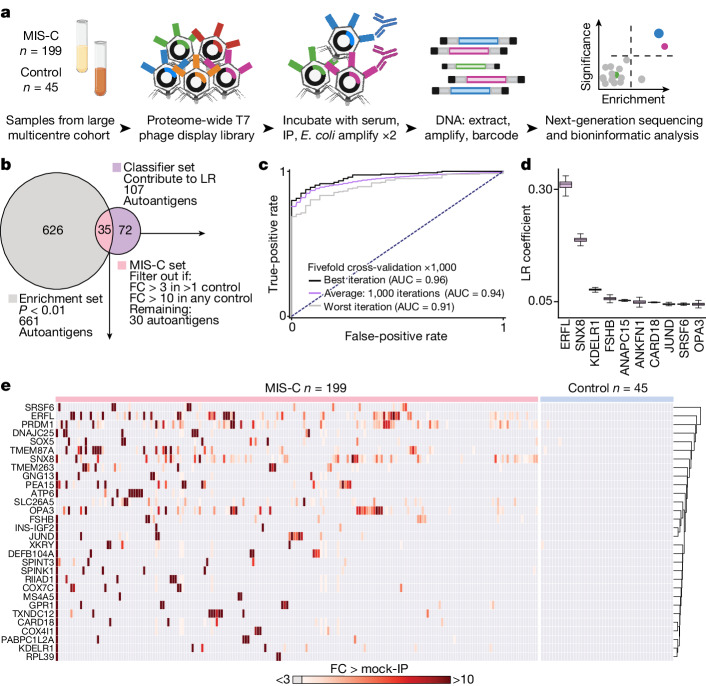
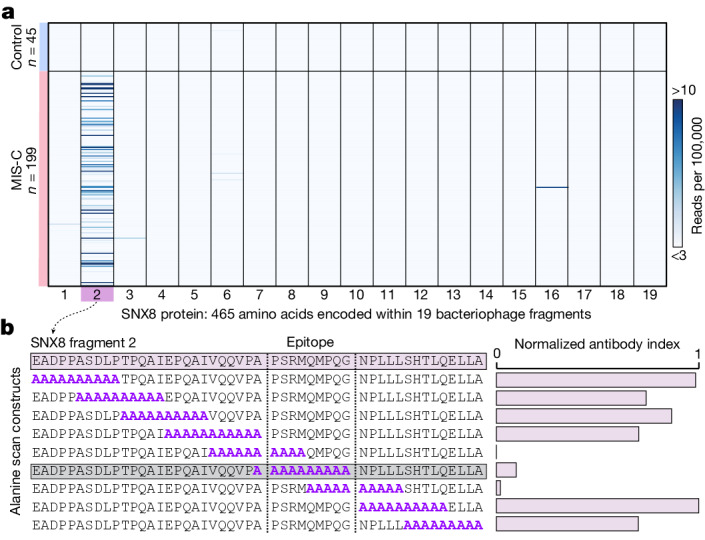
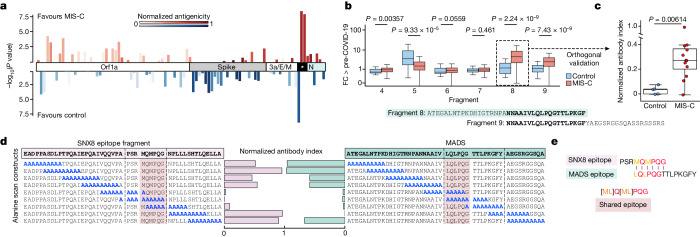
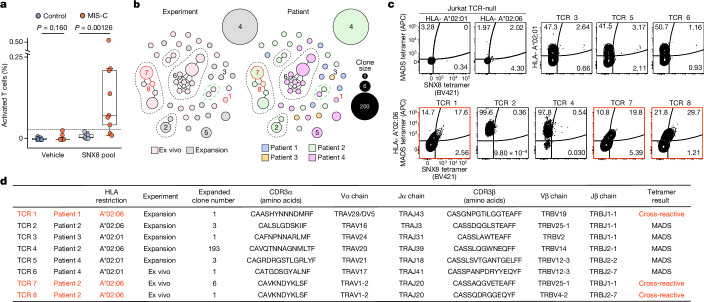
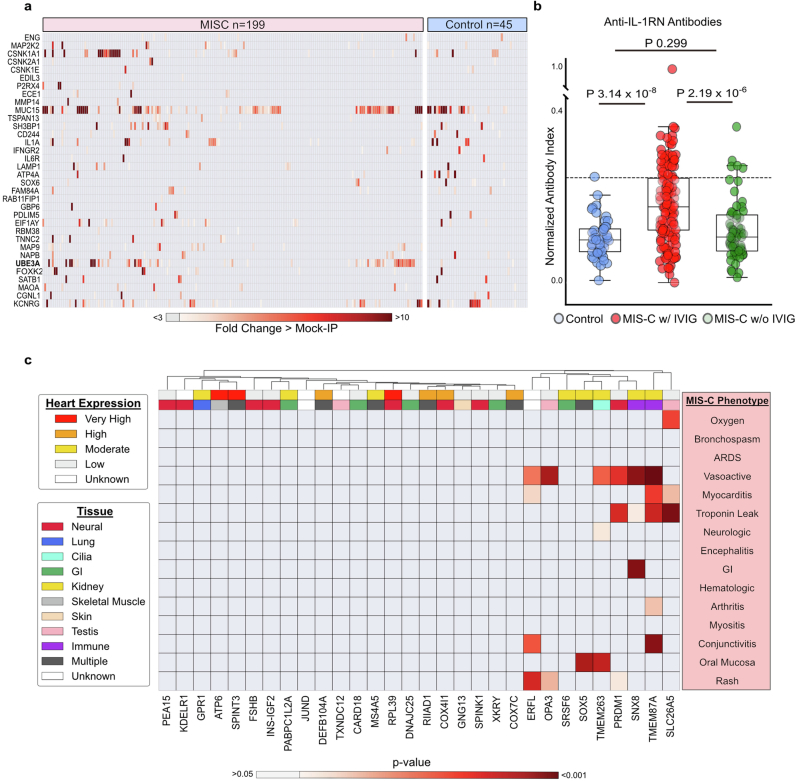
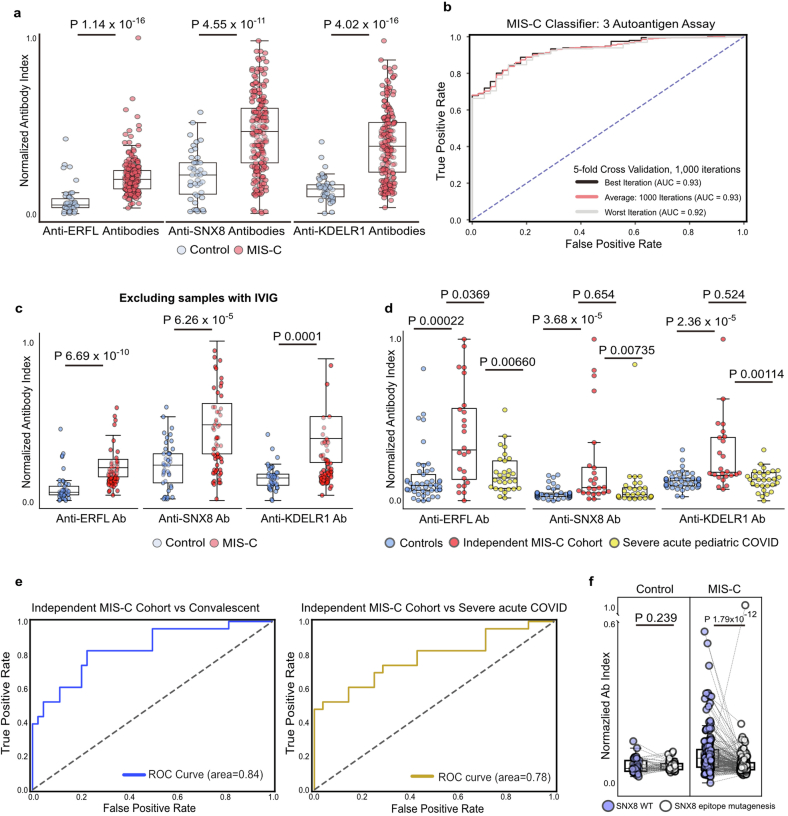
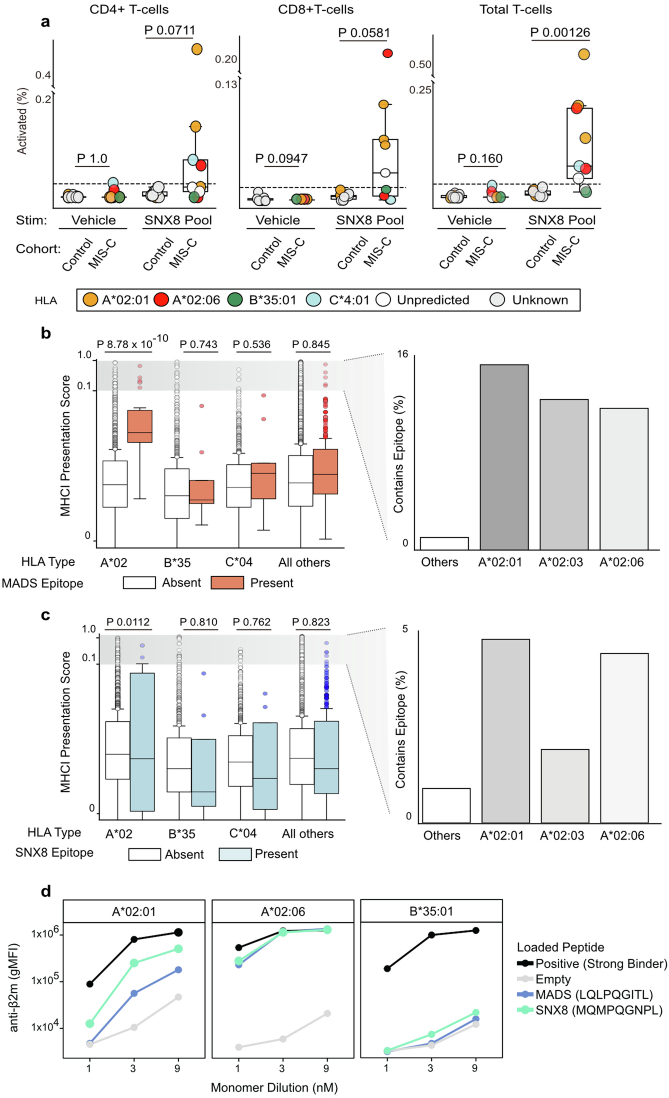
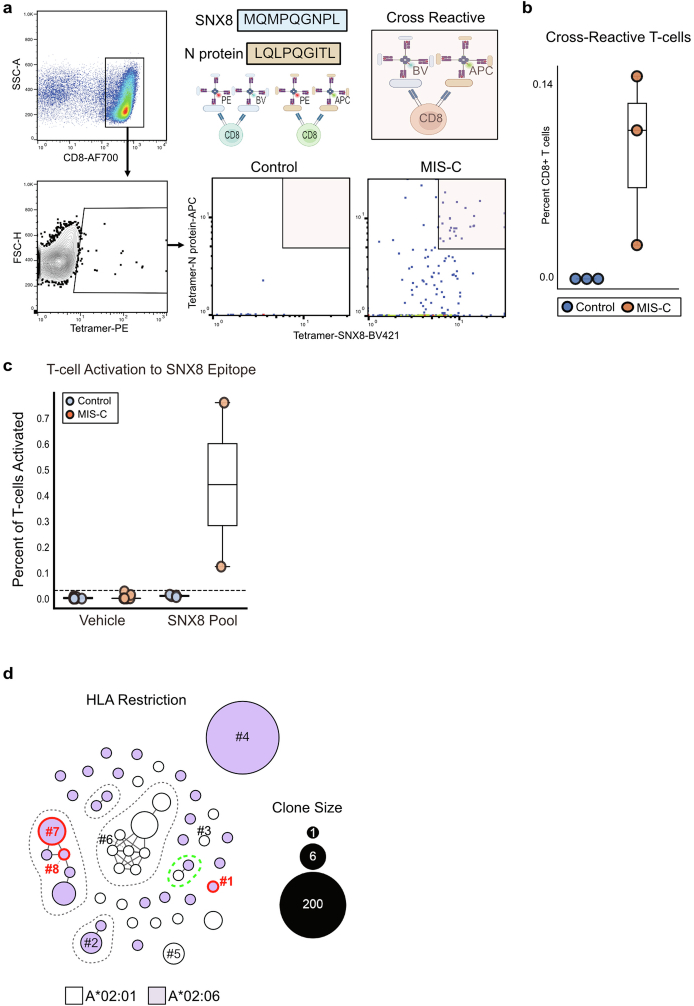
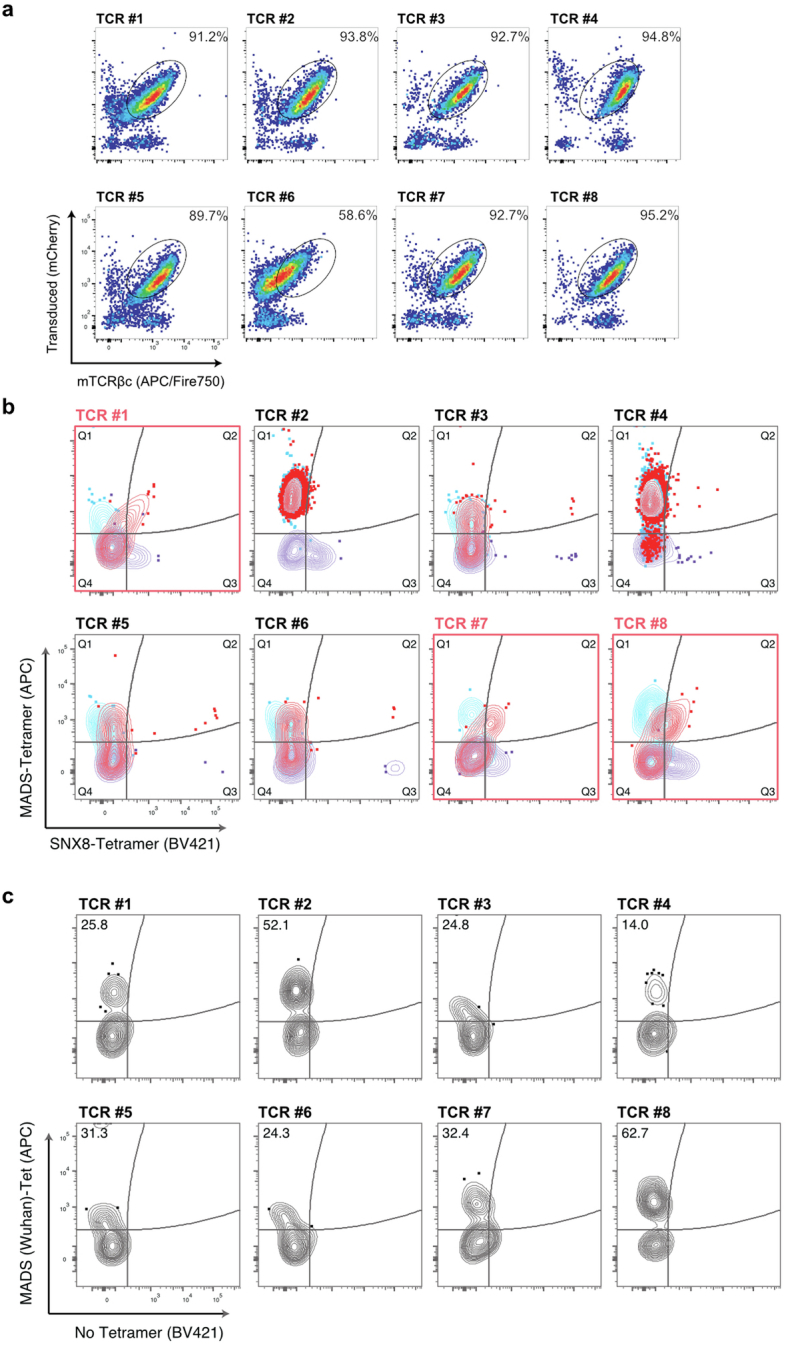
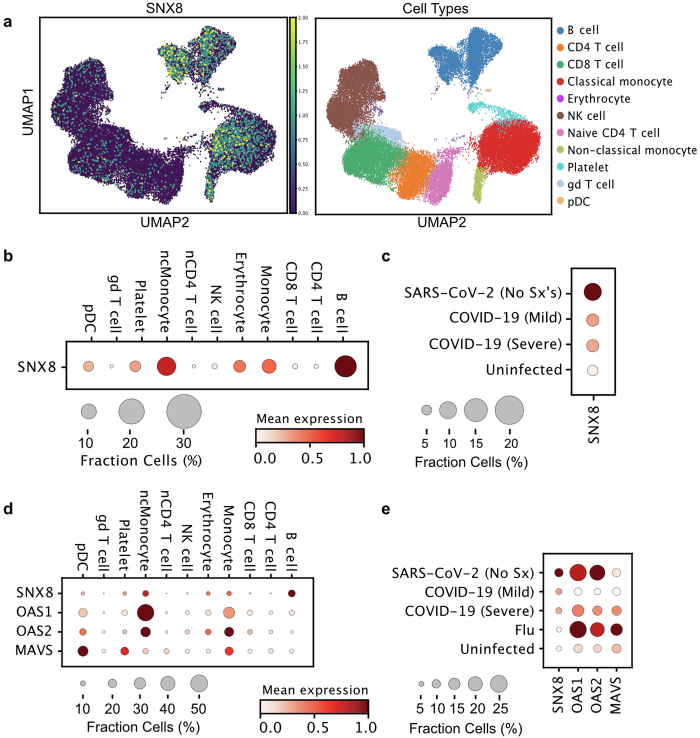
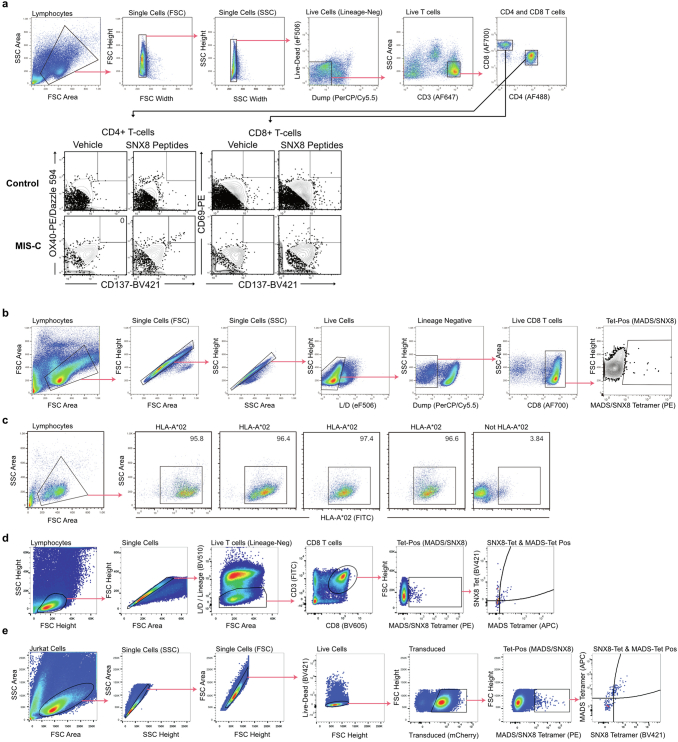
Multisystem inflammatory syndrome in children (MIS-C) is a severe, post-infectious sequela of SARS-CoV-2 infection1,2, yet the pathophysiological mechanism connecting the infection to the broad inflammatory syndrome remains unknown. Here we leveraged a large set of samples from patients with MIS-C to identify a distinct set of host proteins targeted by patient autoantibodies including a particular autoreactive epitope within SNX8, a protein involved in regulating an antiviral pathway associated with MIS-C pathogenesis. In parallel, we also probed antibody responses from patients with MIS-C to the complete SARS-CoV-2 proteome and found enriched reactivity against a distinct domain of the SARS-CoV-2 nucleocapsid protein. The immunogenic regions of the viral nucleocapsid and host SNX8 proteins bear remarkable sequence similarity. Consequently, we found that many children with anti-SNX8 autoantibodies also have cross-reactive T cells engaging both the SNX8 and the SARS-CoV-2 nucleocapsid protein epitopes. Together, these findings suggest that patients with MIS-C develop a characteristic immune response to the SARS-CoV-2 nucleocapsid protein that is associated with cross-reactivity to the self-protein SNX8, demonstrating a mechanistic link between the infection and the inflammatory syndrome, with implications for better understanding a range of post-infectious autoinflammatory diseases.
© 2024. The Author(s).
Conflict of interest statement
J.L.D. reports being a founder and paid consultant for Delve Bio, Inc., and a paid consultant for the Public Health Company and Allen & Co. M.A.S. receives unrelated research funding from the National Institutes of Health, the Centers for Disease Control and Prevention, Cepheid and Merck and unrelated honoria from UpToDate, Inc. M.R.W. receives unrelated research grant funding from Roche/Genentech and Novartis, and received speaking honoraria from Genentech, Takeda, WebMD and Novartis. J.C. reports consulting fees from GLG group, payments from Elsevier for work as an Associate Editor, a patent pending for methods and compositions for treating and preventing T cell-driven diseases, payments related to participation on a Data Safety Monitoring Board or Advisory Board for Enzyvant, and is a member of the Diagnostic Laboratory Immunology Committee of the Clinical Immunology Society. M.S.Z. receives unrelated funding from the National Heart, Lung, and Blood Institute and consults for Sobi. N.B.H. reports unrelated previous grant support from Sanofi and Quidel, and current grant support from Merck. C.V.H. reports being a speaker for Biofire and a reviewer for UpToDate, Inc. and Dynamed.com. A.G.R. receives royalties as a section editor for Pediatric Critical Care Medicine UpToDate, Inc., and received honoraria for MIS-C-related Grand Round Presentations. A.G.R. is also on the medical advisor board of Families Fighting Flu and is Chair of the International Sepsis Forum, which is supported by industry and has received reagents from Illumina, Inc. P.G.T. is on the Scientific Advisory Board of Immunoscape and Shennon Bio, has received research support and personal fees from Elevate Bio, and consulted for 10X Genomics, Illumina, Pfizer, Cytoagents, Merck and JNJ. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention nor the National Institute of Allergy and Infectious Diseases. All other authors declare no competing interests.
Figures
References
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
