

Prognostic genome and transcriptome signatures in colorectal cancers
- PMID: 39112715
- PMCID: PMC11374687
- DOI: 10.1038/s41586-024-07769-3
Prognostic genome and transcriptome signatures in colorectal cancers
Abstract
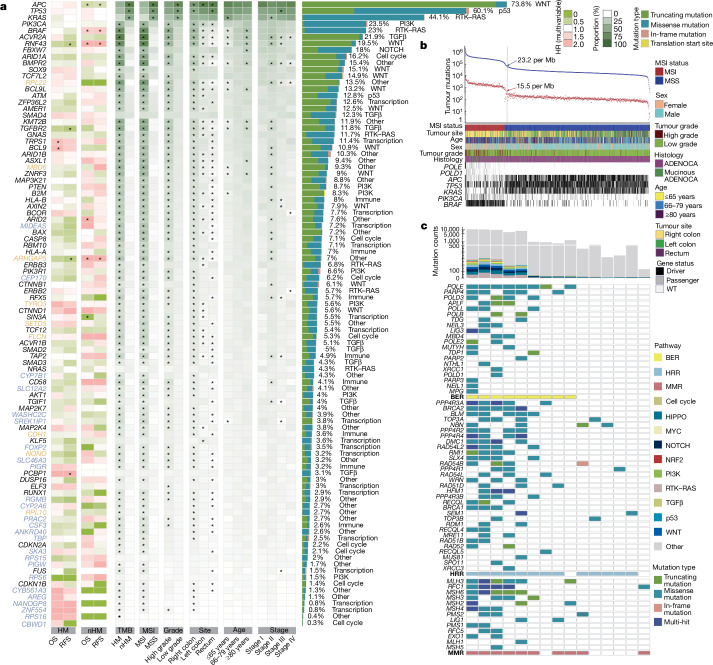
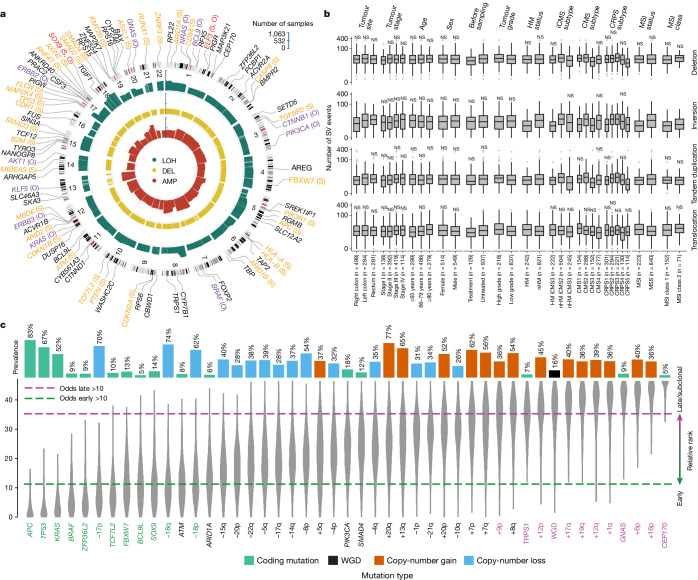
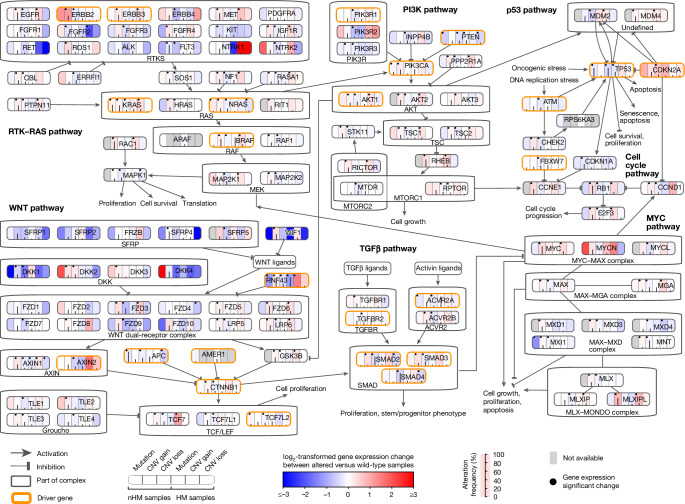
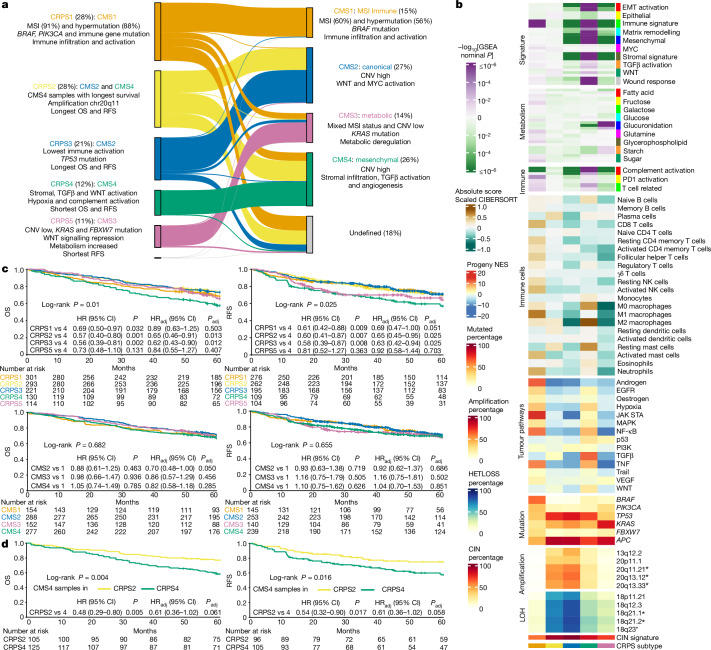
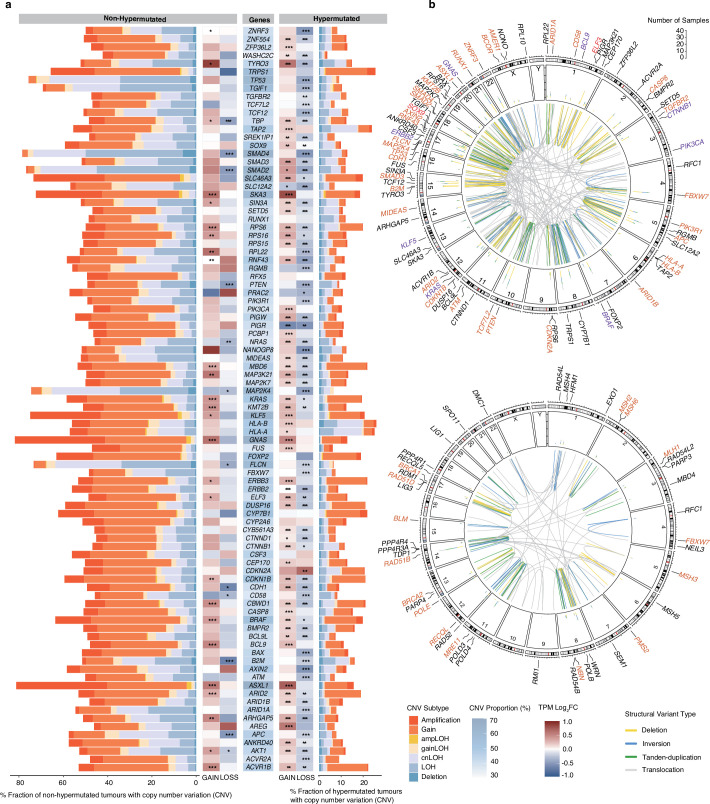
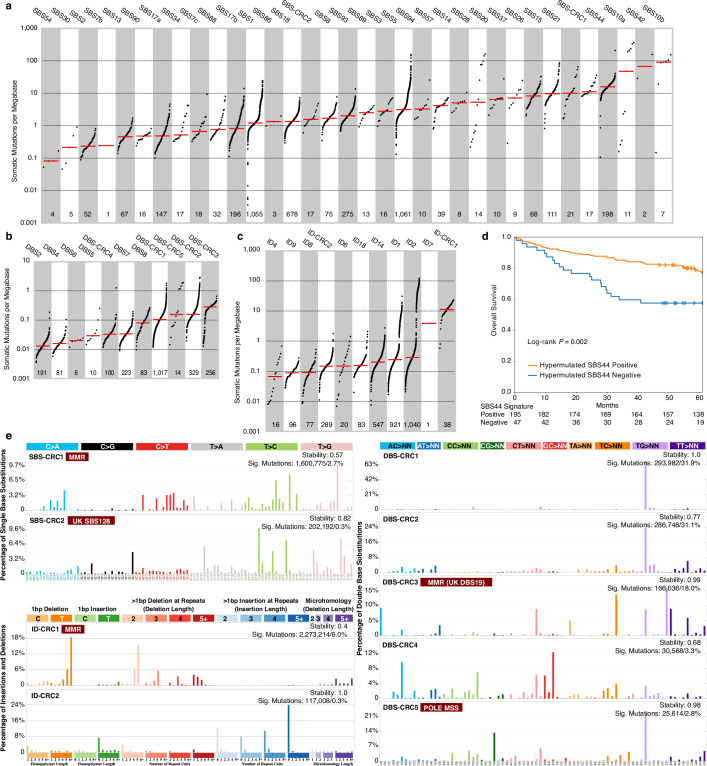
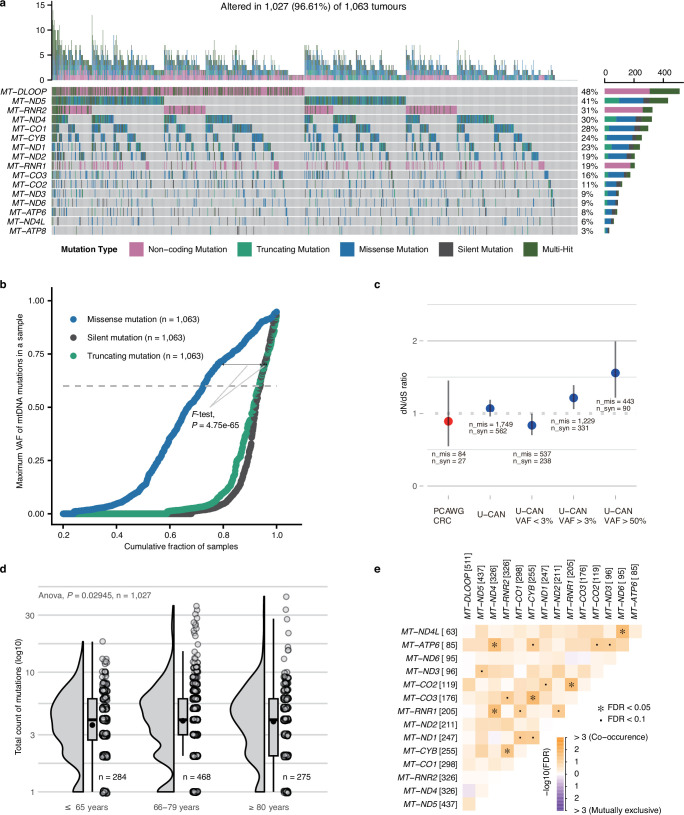
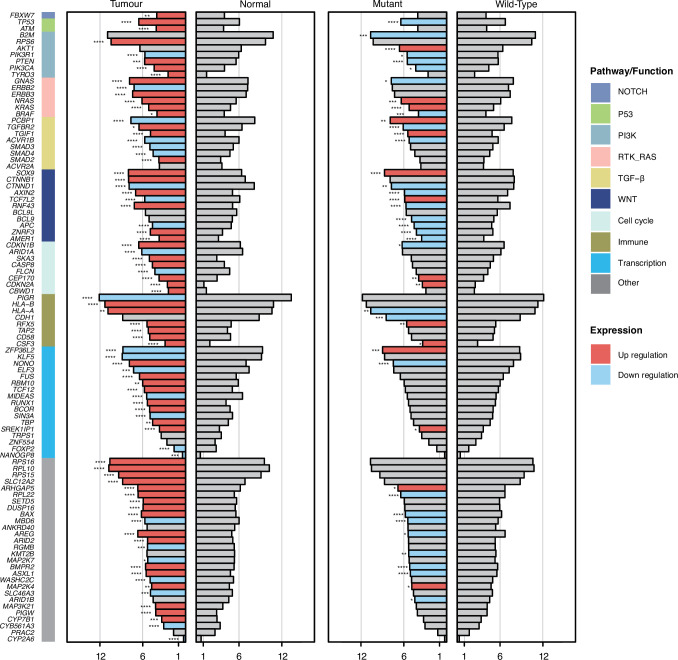
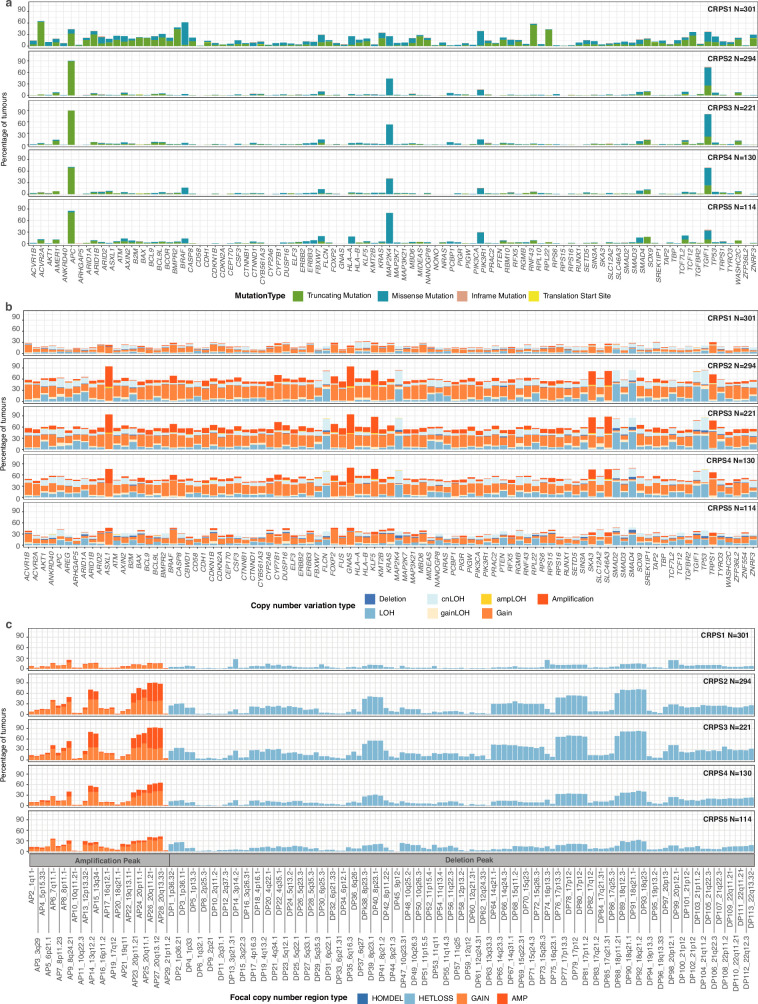
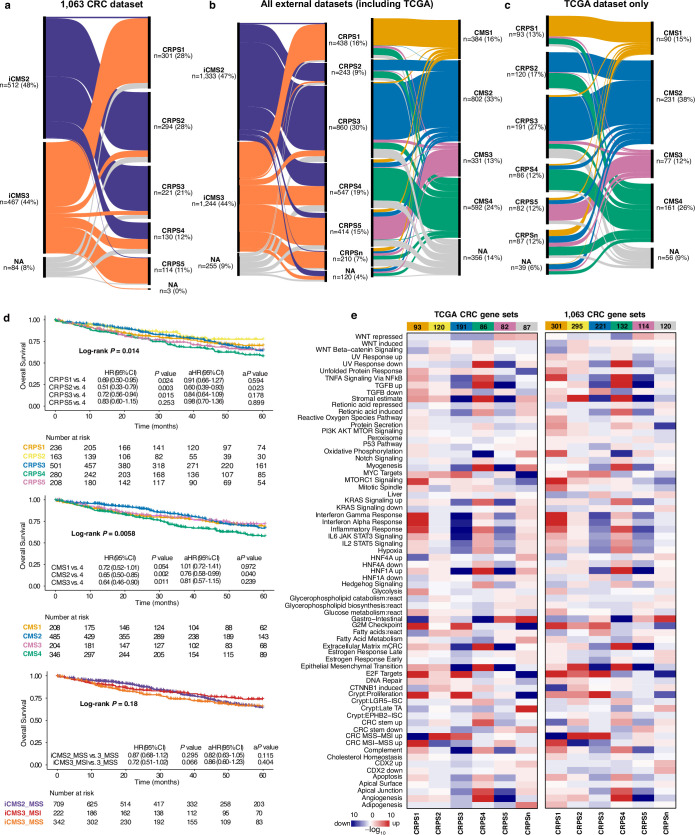
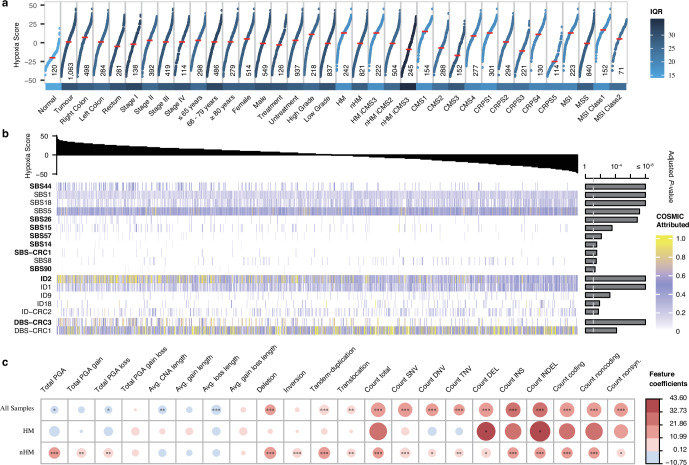
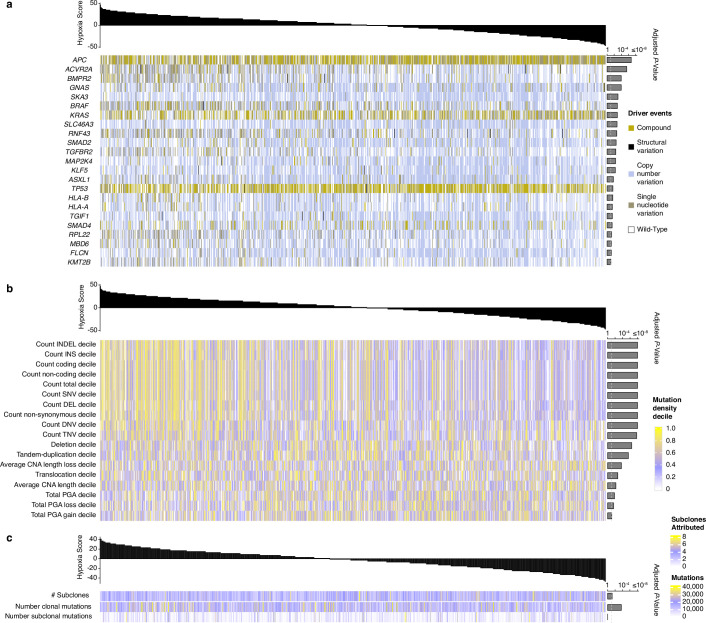
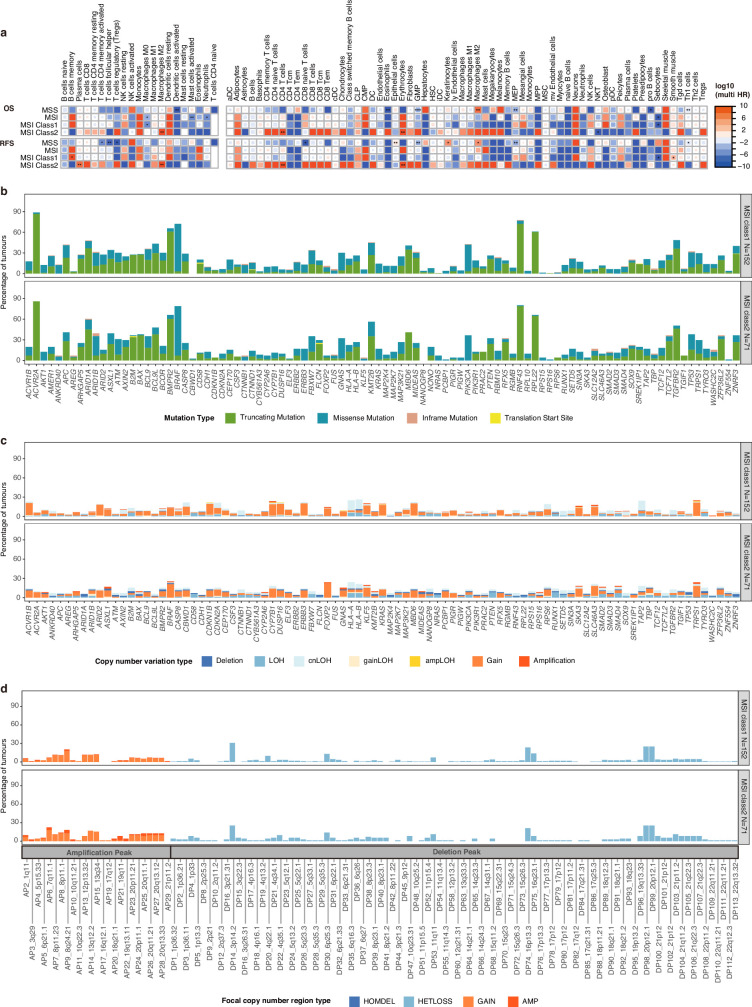
Colorectal cancer is caused by a sequence of somatic genomic alterations affecting driver genes in core cancer pathways1. Here, to understand the functional and prognostic impact of cancer-causing somatic mutations, we analysed the whole genomes and transcriptomes of 1,063 primary colorectal cancers in a population-based cohort with long-term follow-up. From the 96 mutated driver genes, 9 were not previously implicated in colorectal cancer and 24 had not been linked to any cancer. Two distinct patterns of pathway co-mutations were observed, timing analyses identified nine early and three late driver gene mutations, and several signatures of colorectal-cancer-specific mutational processes were identified. Mutations in WNT, EGFR and TGFβ pathway genes, the mitochondrial CYB gene and 3 regulatory elements along with 21 copy-number variations and the COSMIC SBS44 signature correlated with survival. Gene expression classification yielded five prognostic subtypes with distinct molecular features, in part explained by underlying genomic alterations. Microsatellite-instable tumours divided into two classes with different levels of hypoxia and infiltration of immune and stromal cells. To our knowledge, this study constitutes the largest integrated genome and transcriptome analysis of colorectal cancer, and interlinks mutations, gene expression and patient outcomes. The identification of prognostic mutations and expression subtypes can guide future efforts to individualize colorectal cancer therapy.
© 2024. The Author(s).
Conflict of interest statement
Authors declare no competing interests.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
