

Distinctive genomic features of human T-lymphotropic virus type 1-related adult T-cell leukemia-lymphoma in Western populations
- PMID: 39113648
- PMCID: PMC11609816
- DOI: 10.3324/haematol.2024.285233
Distinctive genomic features of human T-lymphotropic virus type 1-related adult T-cell leukemia-lymphoma in Western populations
Abstract
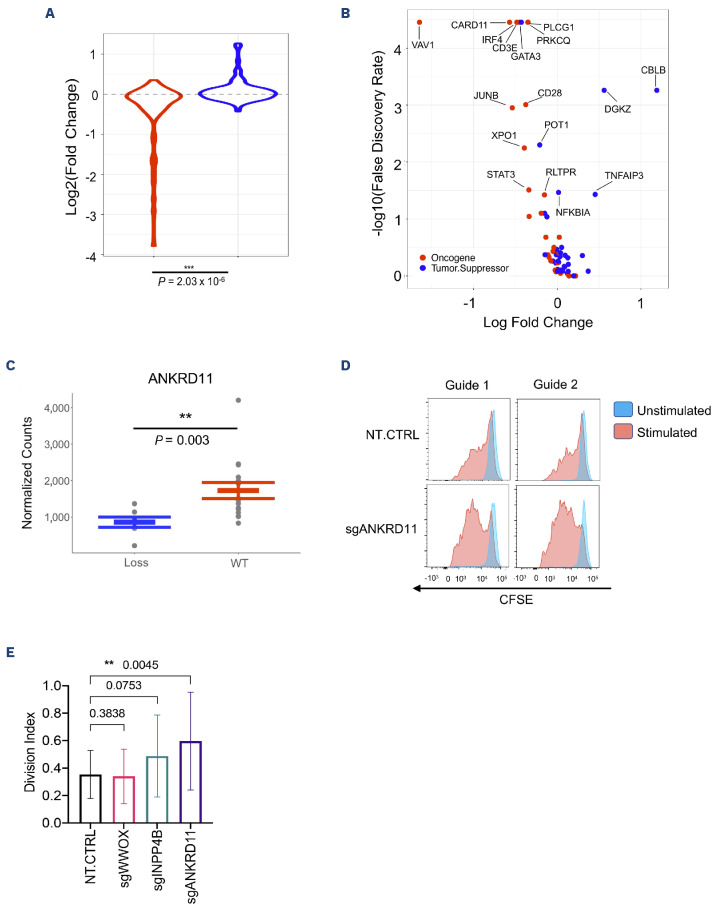
Adult T-cell leukemia-lymphoma (ATLL) is an aggressive malignancy driven by human T-cell leukemia virus type 1 (HTLV-1). Although patients from the Western hemisphere (Afro-Caribbean and South American) face worse prognoses, our understanding of ATLL molecular drivers derives mostly from Japanese studies. We performed multi-omic analyses to elucidate the genomic landscape of ATLL in Western cohorts. Recurrent deletions and/or damaging mutations involving FOXO3, ANKRD11, DGKZ, and PTPN6 implicate these genes as potential tumor suppressors. RNA-sequencing, published functional data and in vitro assays support the roles of ANKRD11 and FOXO3 as regulators of T-cell proliferation and apoptosis in ATLL, respectively. Survival data suggest that ANKRD11 mutation may confer a worse prognosis. Japanese and Western cohorts, in addition to acute and lymphomatous subtypes, demonstrated distinct molecular patterns. GATA3 deletion was associated with chronic cases with unfavorable outcomes. IRF4 and CARD11 mutations frequently emerged in relapses after interferon therapy. Our findings reveal novel putative ATLL driver genes and clinically relevant differences between Japanese and Western ATLL patients.
Figures






References
-
- Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984-87). Br J Haematol. 1991;79(3):428-437. - PubMed
-
- Ishitsuka K, Tamura K. Human T-cell leukaemia virus type I and adult T-cell leukaemia-lymphoma. Lancet Oncol. 2014;15(11):e517-526. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
