

Cellular and functional evaluation of LDLR missense variants reported in hypercholesterolemic patients demonstrates their hypomorphic impacts on trafficking and LDL internalization
- PMID: 39114568
- PMCID: PMC11303217
- DOI: 10.3389/fcell.2024.1412236
Cellular and functional evaluation of LDLR missense variants reported in hypercholesterolemic patients demonstrates their hypomorphic impacts on trafficking and LDL internalization
Abstract
Background: Familial hypercholesterolemia (FH) is an autosomal dominant disorder characterized by increased LDL-cholesterol levels. About 85% of FH cases are caused by LDLR mutations encoding the low-density lipoprotein receptor (LDLR). LDLR is synthesized in the endoplasmic reticulum (ER) where it undergoes post-translational modifications and then transported through Golgi apparatus to the plasma membrane. Over 2900 LDLR variants have been reported in FH patients with limited information on the pathogenicity and functionality of many of them. This study aims to elucidate the cellular trafficking and functional implications of LDLR missense variants identified in suspected FH patients using biochemical and functional methods.
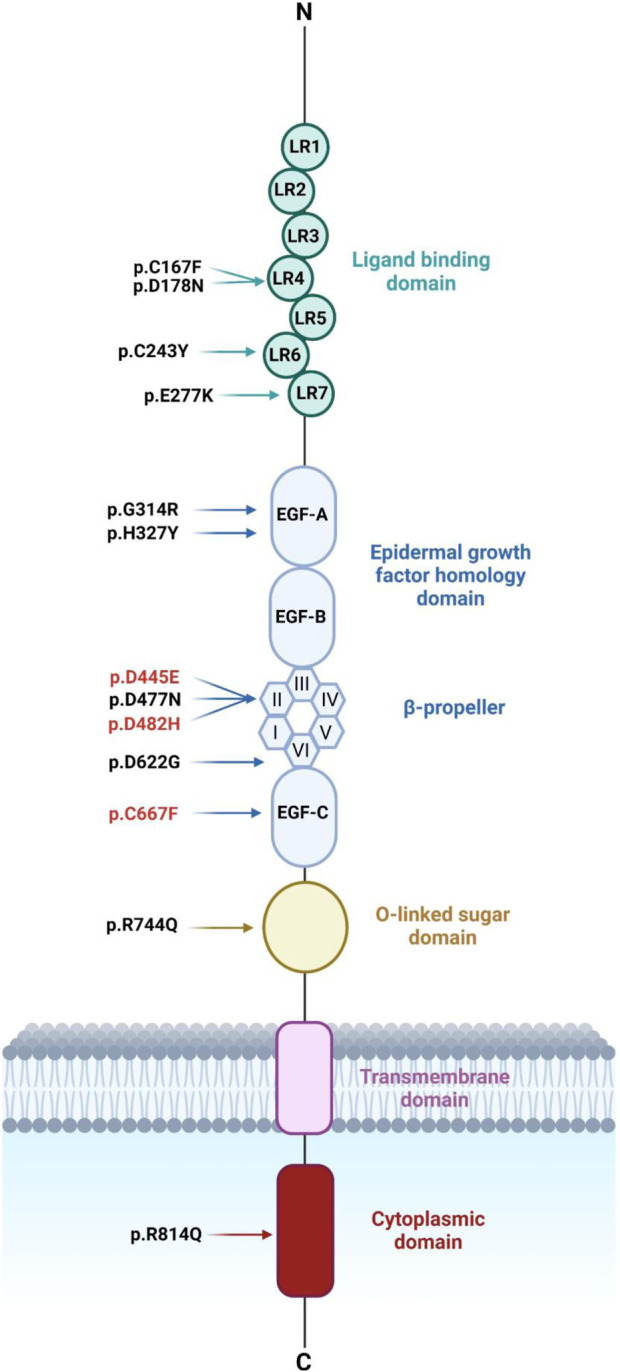
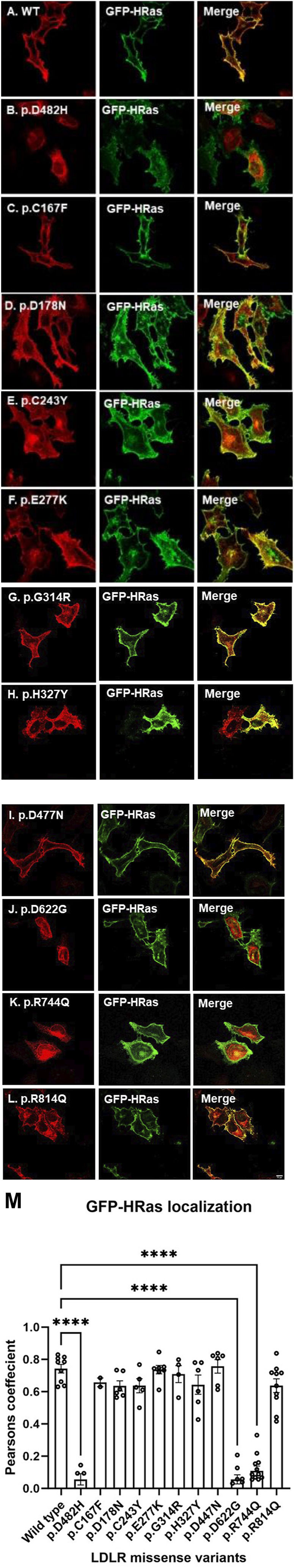
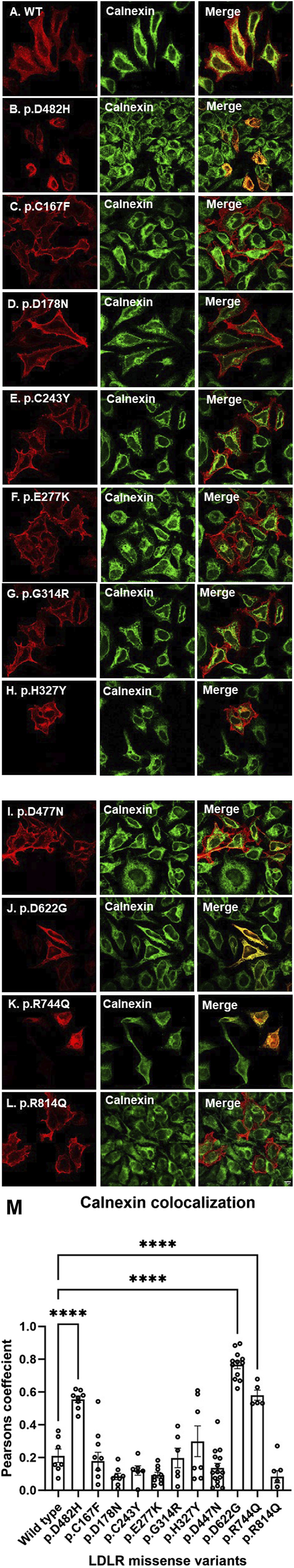
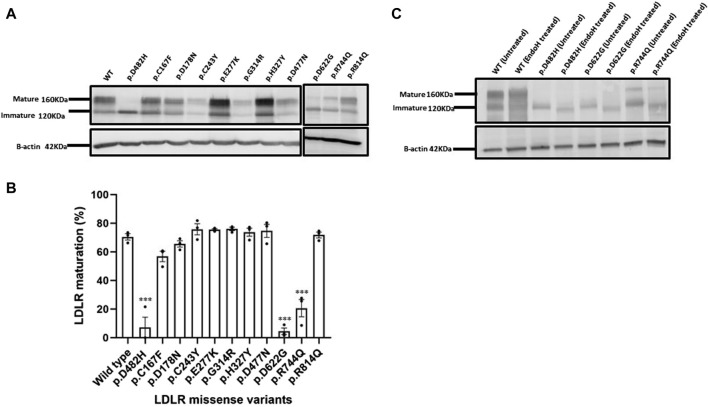
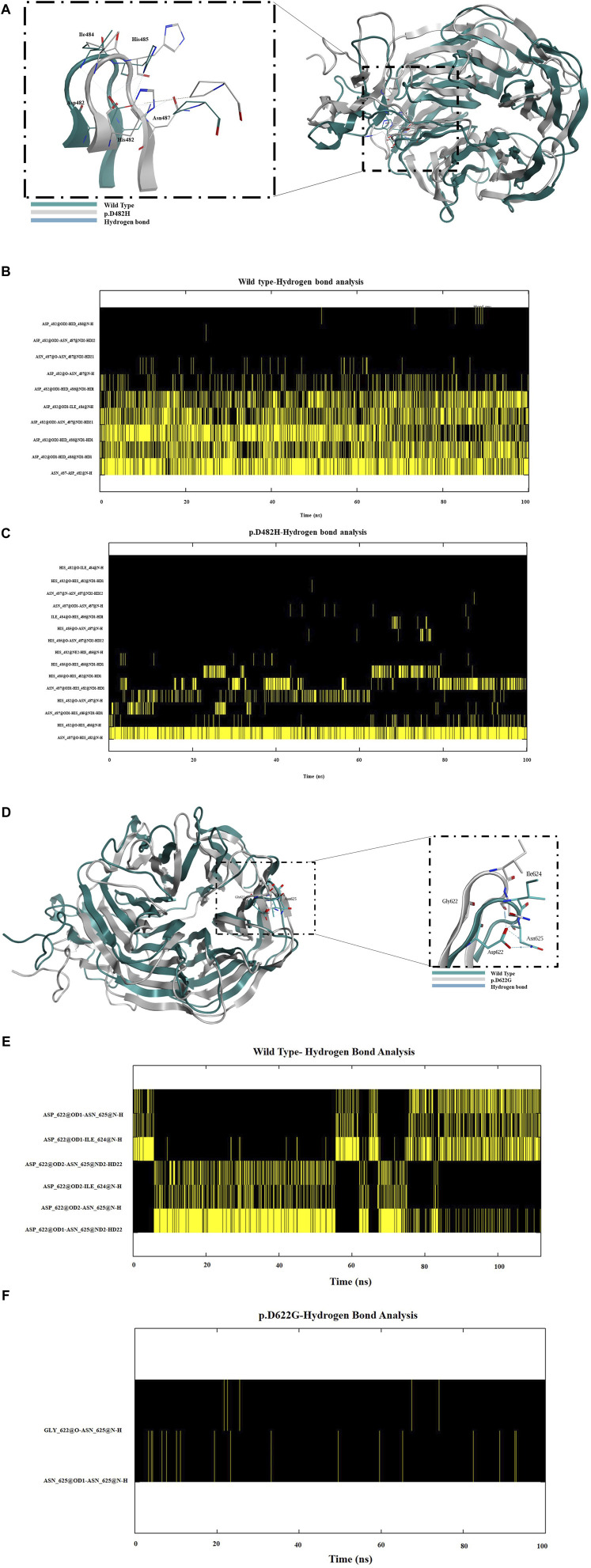
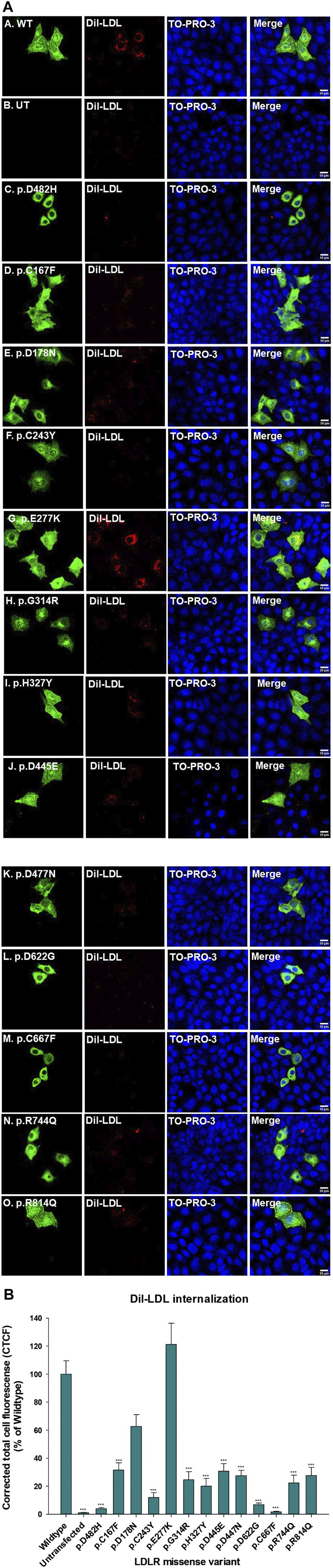
Methods: We used HeLa, HEK293T, and LDLR-deficient-CHO-ldlA7 cells to evaluate the subcellular localization and LDL internalization of ten LDLR missense variants (p.C167F, p.D178N, p.C243Y, p.E277K, p.G314R, p.H327Y, p.D477N, p.D622G, p.R744Q, and p.R814Q) reported in multiethnic suspected FH patients. We also analyzed the functional impact of three variants (p.D445E, p.D482H, and p.C677F), two of which previously shown to be retained in the ER.
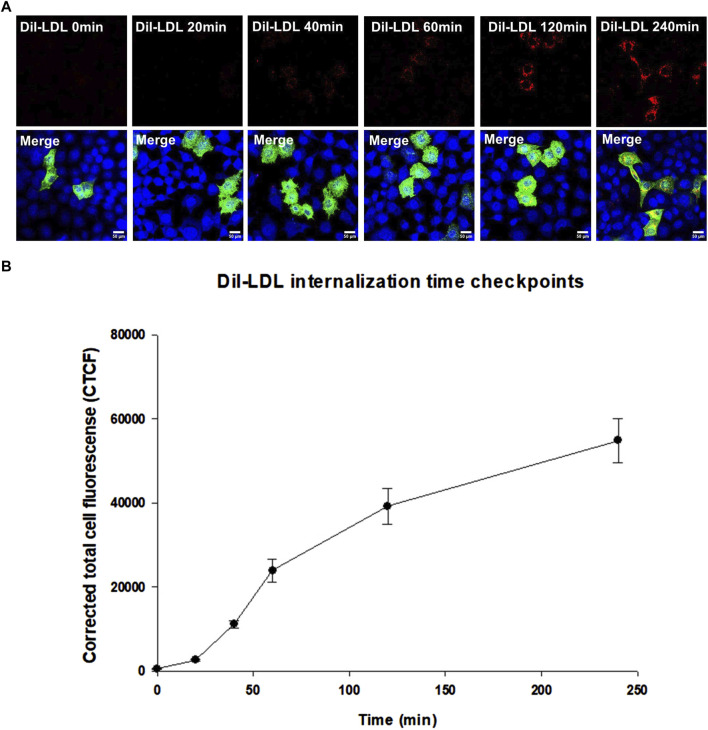
Results: We show that p.D622G, p.D482H, and p.C667F are largely retained in the ER whereas p.R744Q is partially retained. The other variants were predominantly localized to the plasma membrane. LDL internalization assays in CHO-ldlA7 cells indicate that p.D482H, p.C243Y, p.D622G, and p.C667F have quantitatively lost their ability to internalize Dil-LDL with the others (p.C167F, p.D178N, p.G314R, p.H327Y, p.D445E, p.D477N, p.R744Q and p.R814Q) showing significant losses except for p.E277K which retained full activity. However, the LDL internalization assay is only to able evaluate the impact of the variants on LDL internalization and not the exact functional defects such as failure to bind LDL. The data represented illustrate the hypomorphism nature of variants causing FH which may explain some of the variable expressivity of FH.
Conclusion: Our combinatorial approach of in silico, cellular, and functional analysis is a powerful strategy to determine pathogenicity and FH disease mechanisms which may provide opportunitites for novel therapeutic strategies.
Keywords: ER associated protein degradation (ERAD); ER stress; familial hypercholesterolemia (FH); low-density lipoprotein (LDL); low-density lipoprotein receptor (LDLR); protein quality control; receptor-mediated endocytosis.
Copyright © 2024 Jawabri, John, Ghattas, Mahgoub, Hamad, Barakat, Shobi, Daggag and Ali.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Arias-Moreno X., Velazquez-Campoy A., Rodríguez J. C., Pocoví M., Sancho J. (2008). Mechanism of low density lipoprotein (LDL) release in the endosome: implications of the stability and Ca2+ affinity of the fifth binding module of the LDL receptor. J. Biol. Chem. 283 (33), 22670–22679. 10.1074/jbc.M802153200 - DOI - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous