

Revisiting Dominance in Population Genetics
- PMID: 39114967
- PMCID: PMC11306932
- DOI: 10.1093/gbe/evae147
Revisiting Dominance in Population Genetics
Abstract
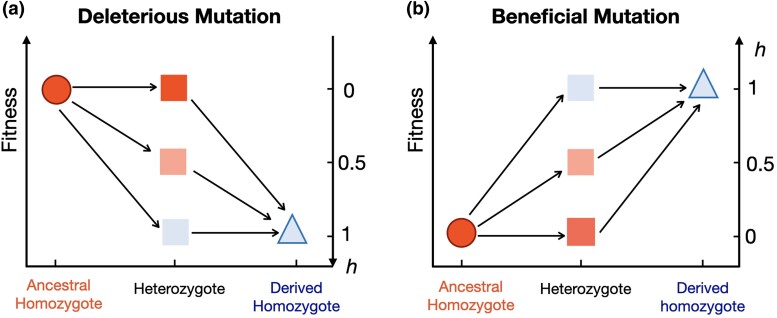
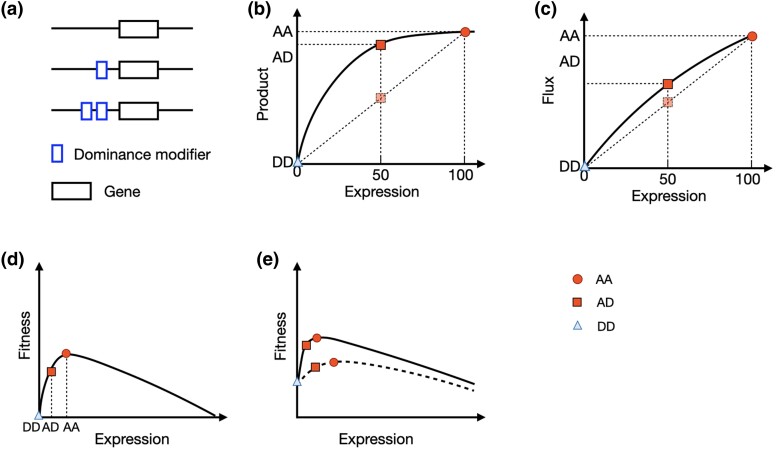
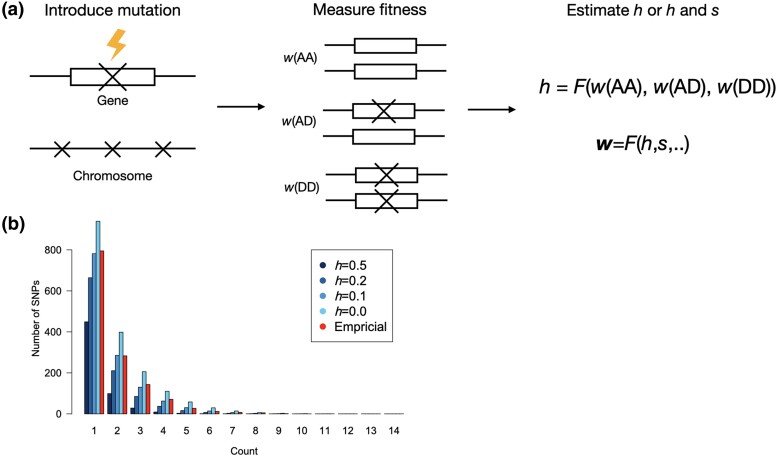
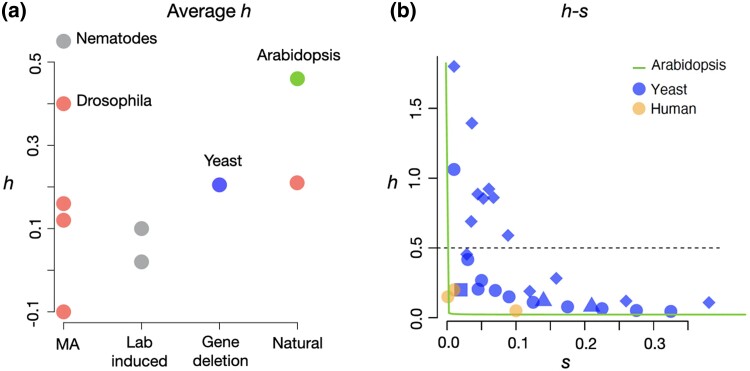
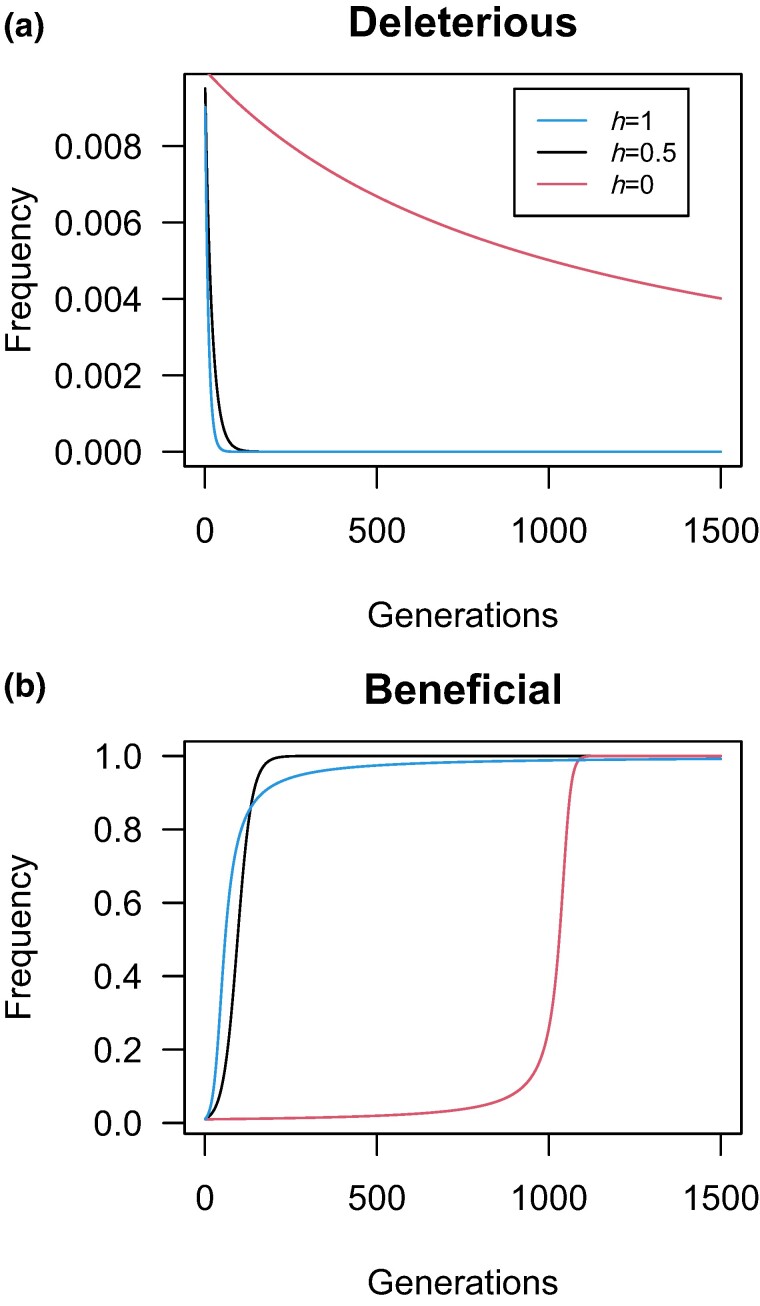
Dominance refers to the effect of a heterozygous genotype relative to that of the two homozygous genotypes. The degree of dominance of mutations for fitness can have a profound impact on how deleterious and beneficial mutations change in frequency over time as well as on the patterns of linked neutral genetic variation surrounding such selected alleles. Since dominance is such a fundamental concept, it has received immense attention throughout the history of population genetics. Early work from Fisher, Wright, and Haldane focused on understanding the conceptual basis for why dominance exists. More recent work has attempted to test these theories and conceptual models by estimating dominance effects of mutations. However, estimating dominance coefficients has been notoriously challenging and has only been done in a few species in a limited number of studies. In this review, we first describe some of the early theoretical and conceptual models for understanding the mechanisms for the existence of dominance. Second, we discuss several approaches used to estimate dominance coefficients and summarize estimates of dominance coefficients. We note trends that have been observed across species, types of mutations, and functional categories of genes. By comparing estimates of dominance coefficients for different types of genes, we test several hypotheses for the existence of dominance. Lastly, we discuss how dominance influences the dynamics of beneficial and deleterious mutations in populations and how the degree of dominance of deleterious mutations influences the impact of inbreeding on fitness.
Keywords: deleterious mutations; dominance; inference; natural selection; population genetics.
© The Author(s) 2024. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures
References
-
- Abu-Awad D, Waller D. Conditions for maintaining and eroding pseudo-overdominance and its contribution to inbreeding depression. Peer Community J. 2023:3:e8. 10.24072/pcjournal.224. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
