

De novo gene synthesis by an antiviral reverse transcriptase
- PMID: 39116258
- PMCID: PMC11758365
- DOI: 10.1126/science.adq0876
De novo gene synthesis by an antiviral reverse transcriptase
Abstract
Defense-associated reverse transcriptase (DRT) systems perform DNA synthesis to protect bacteria against viral infection, but the identities and functions of their DNA products remain largely unknown. We show that DRT2 systems encode an unprecedented immune pathway that involves de novo gene synthesis through rolling circle reverse transcription of a noncoding RNA (ncRNA). Programmed template jumping on the ncRNA generates a concatemeric cDNA, which becomes double-stranded upon viral infection. This DNA product constitutes a protein-coding, nearly endless open reading frame (neo) gene whose expression leads to potent cell growth arrest, restricting the viral infection. Our work highlights an elegant expansion of genome coding potential through RNA-templated gene creation and challenges conventional paradigms of genetic information encoded along the one-dimensional axis of genomic DNA.
Conflict of interest statement
Competing interests:
Columbia University has filed patent applications related to this work, for which S.H.S., S.T., D.J.Z., G.D.L., T.W., and J.T.G. are inventors. S.H.S. is a cofounder and scientific advisor to Dahlia Biosciences, a scientific advisor to CrisprBits and Prime Medicine, and an equity holder in Dahlia Biosciences and CrisprBits.
Figures
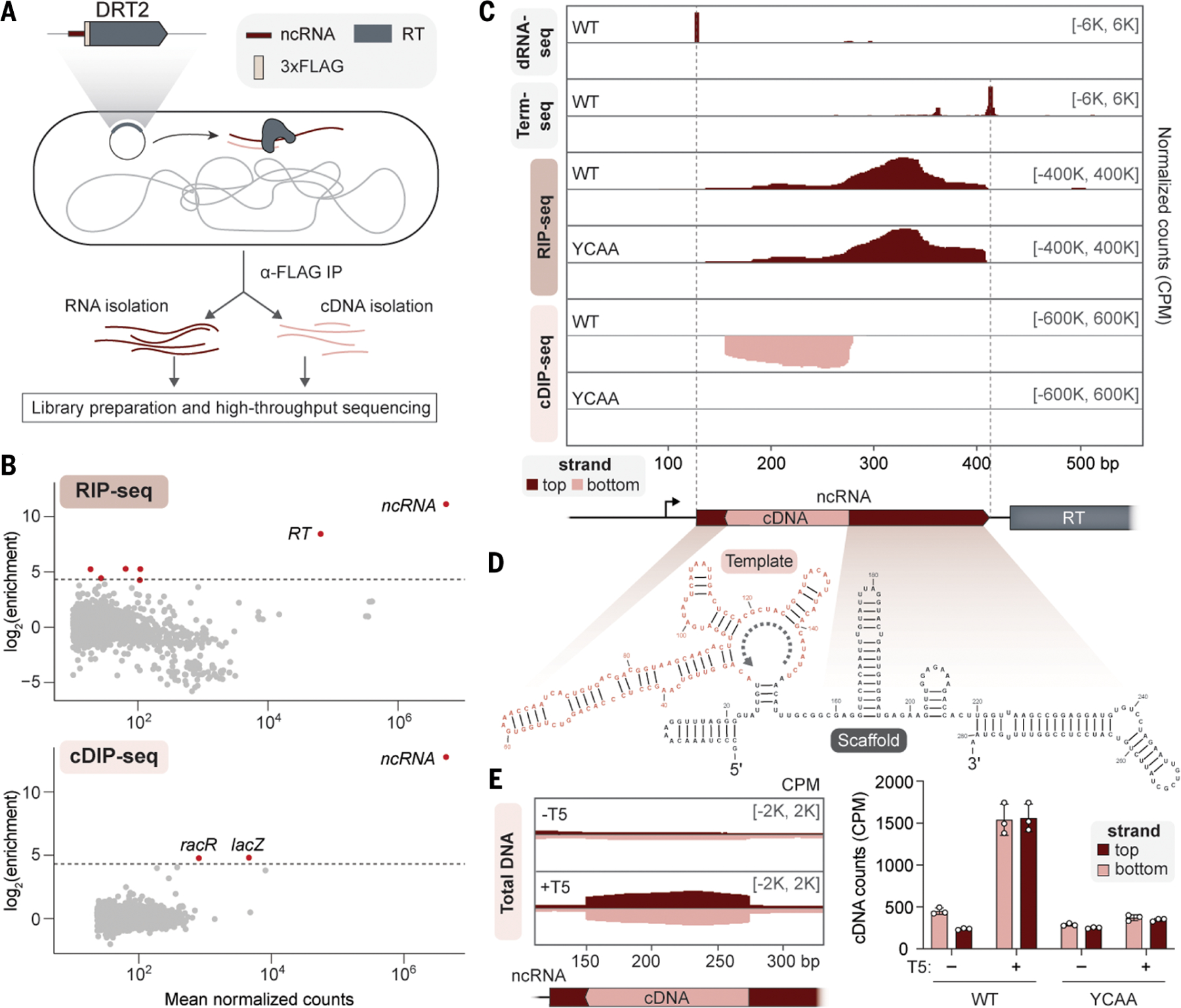
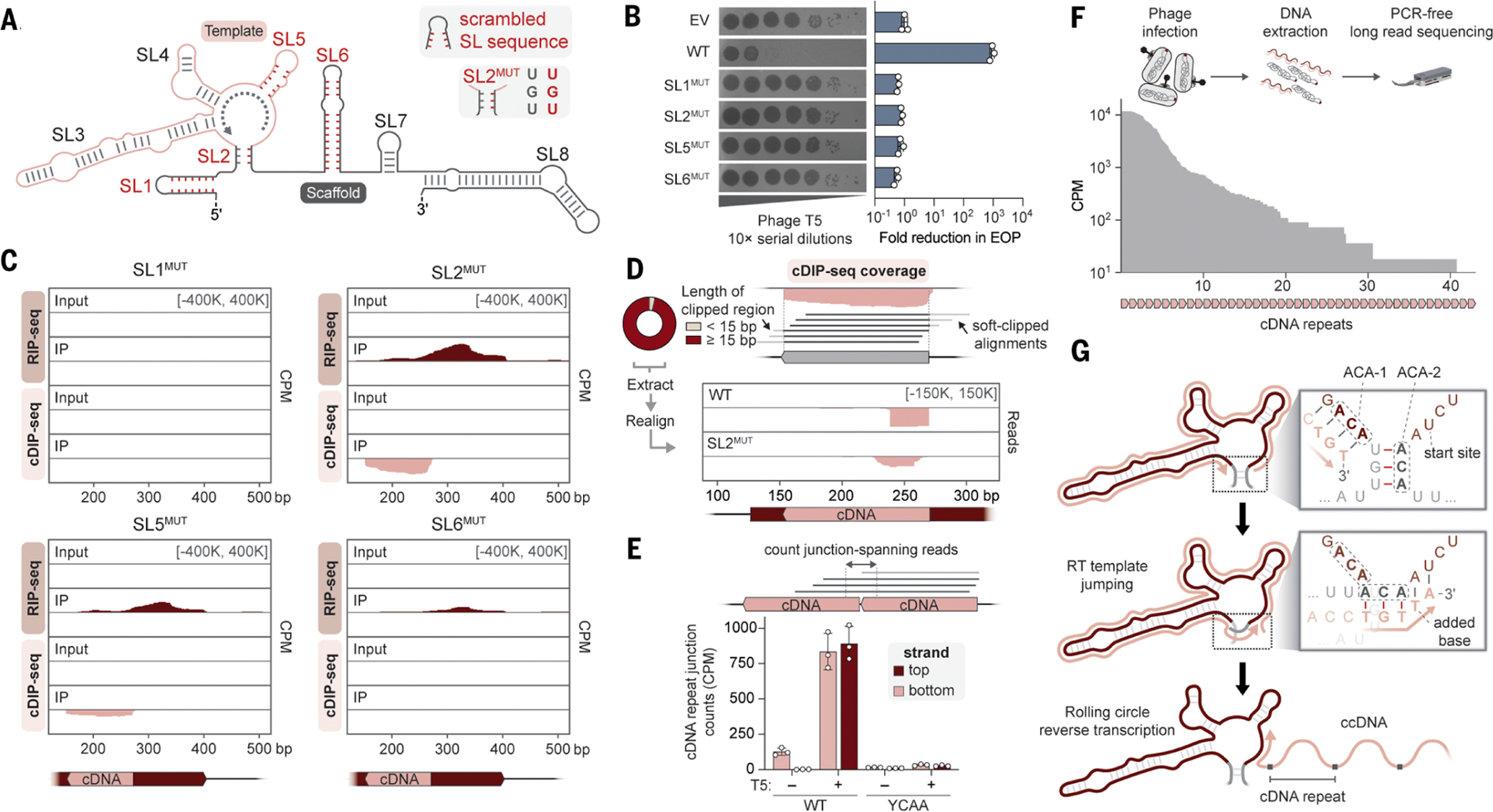



Update of
-
De novo gene synthesis by an antiviral reverse transcriptase.bioRxiv [Preprint]. 2024 May 8:2024.05.08.593200. doi: 10.1101/2024.05.08.593200. bioRxiv. 2024. Update in: Science. 2024 Oct 4;386(6717):eadq0876. doi: 10.1126/science.adq0876. PMID: 38766058 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
