

Single cell dual-omic atlas of the human developing retina
- PMID: 39117640
- PMCID: PMC11310509
- DOI: 10.1038/s41467-024-50853-5
Single cell dual-omic atlas of the human developing retina
Abstract
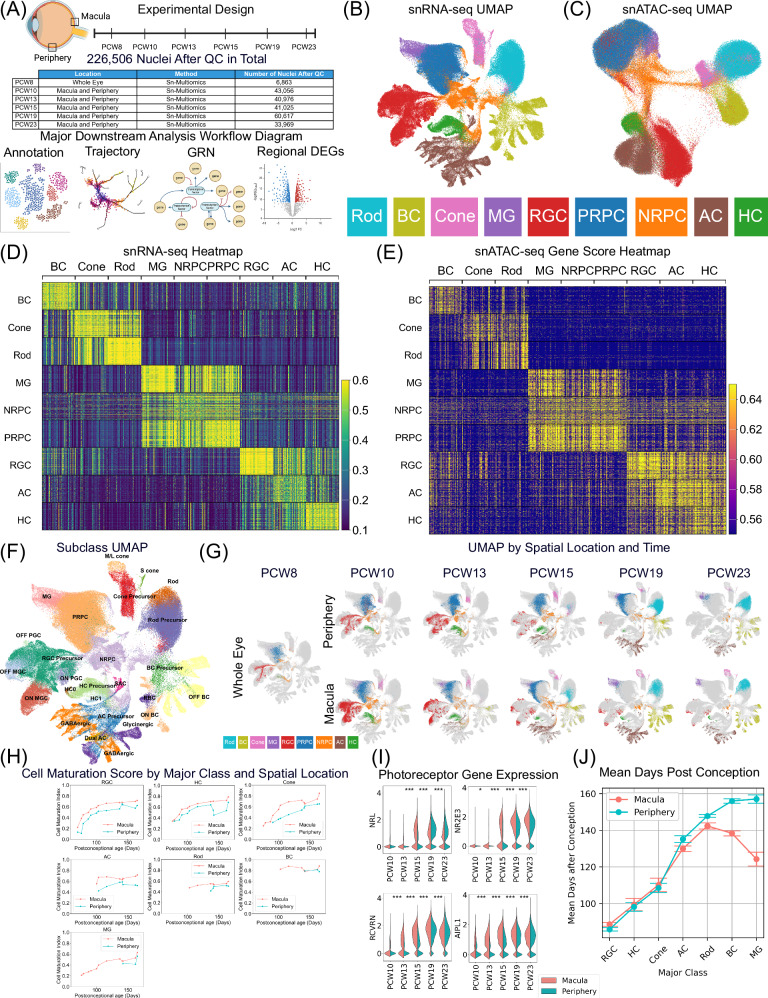
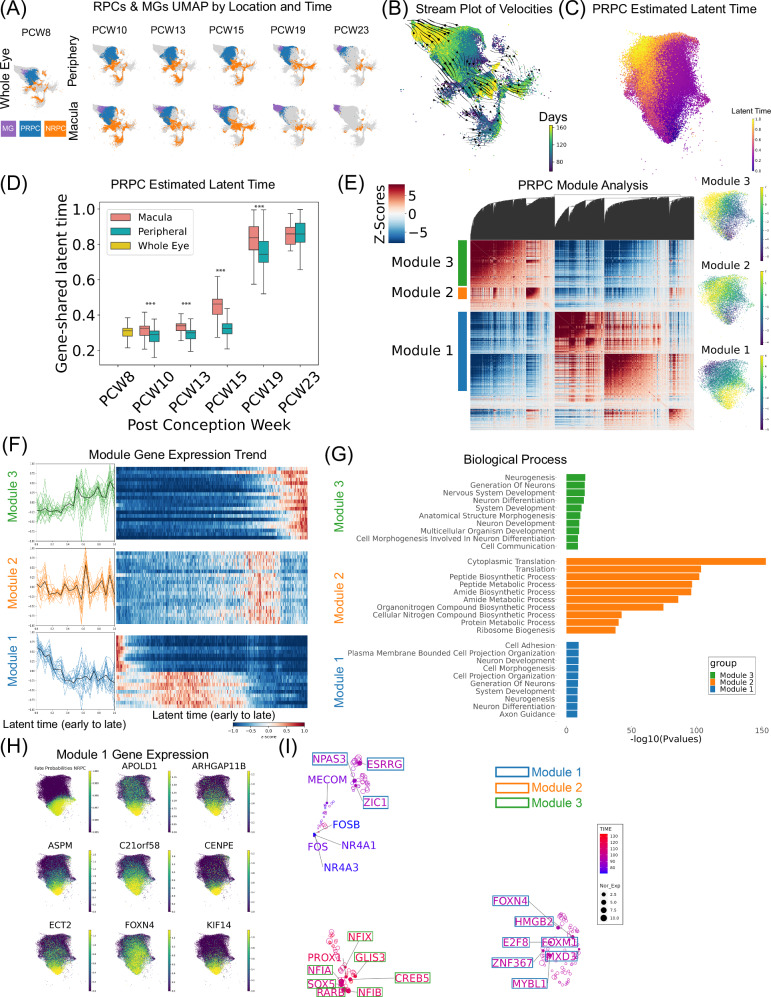
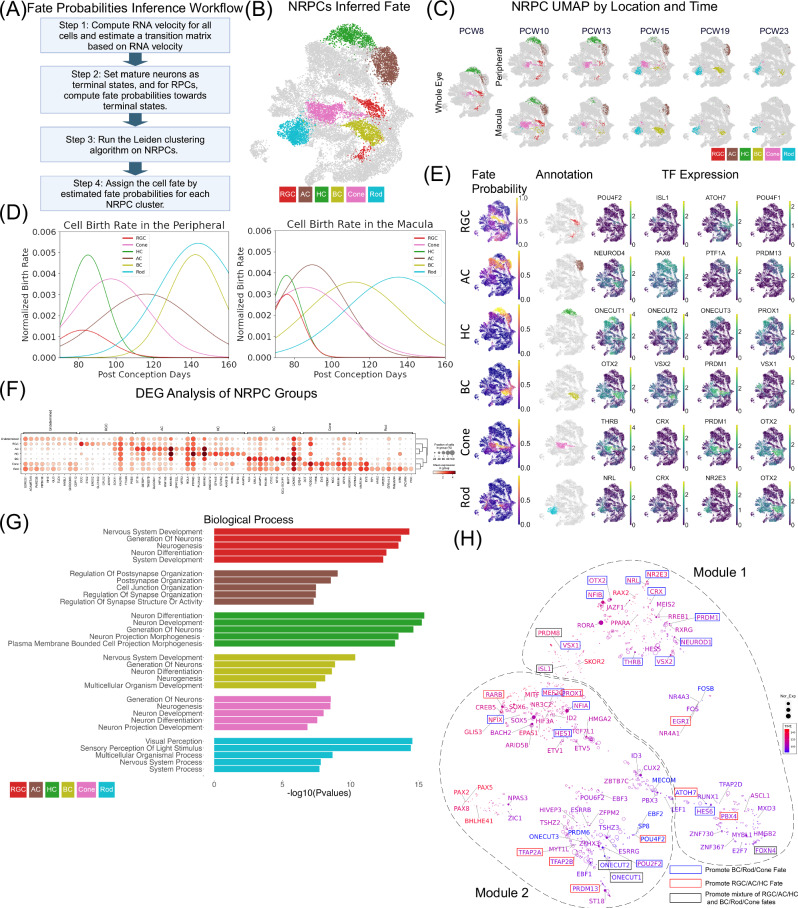
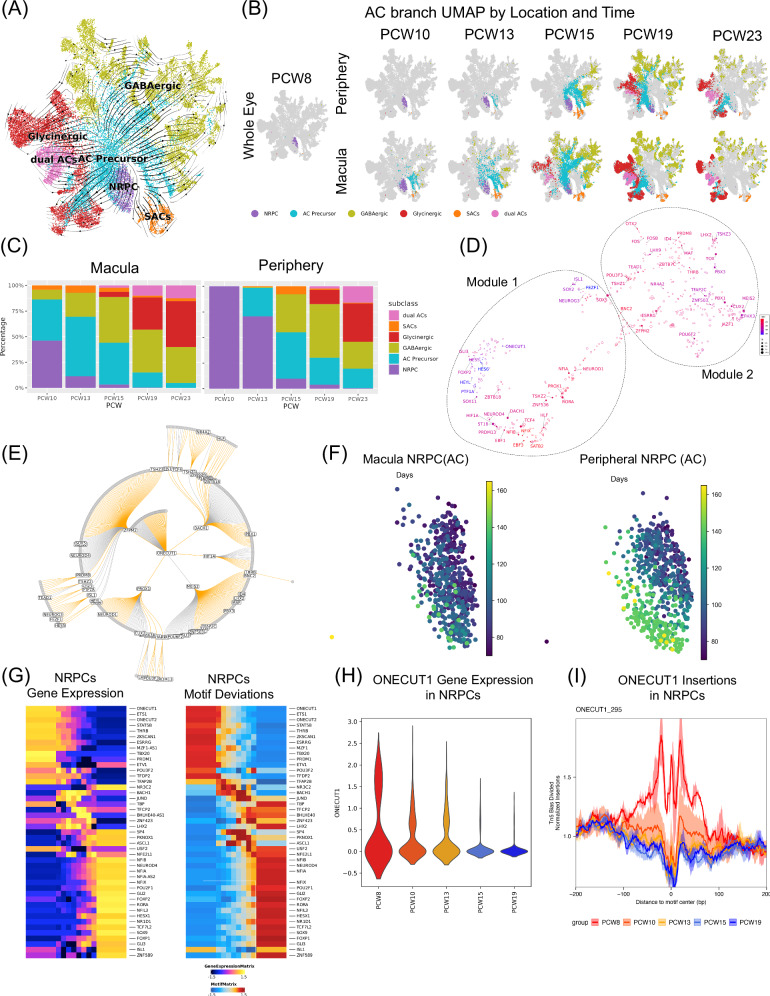
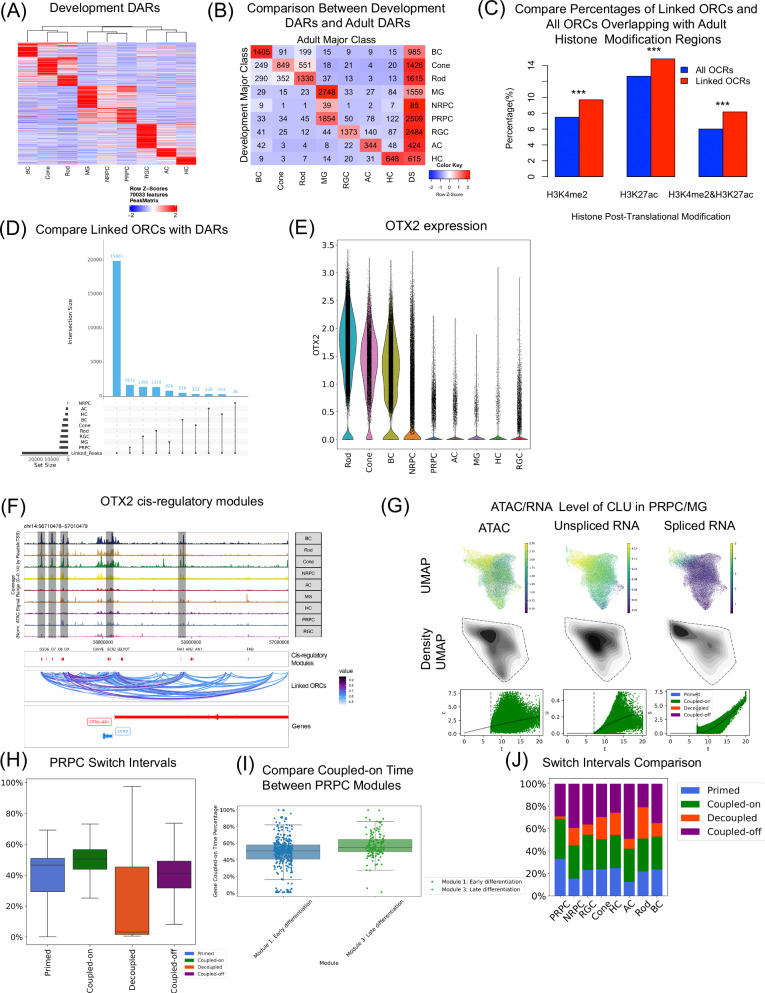
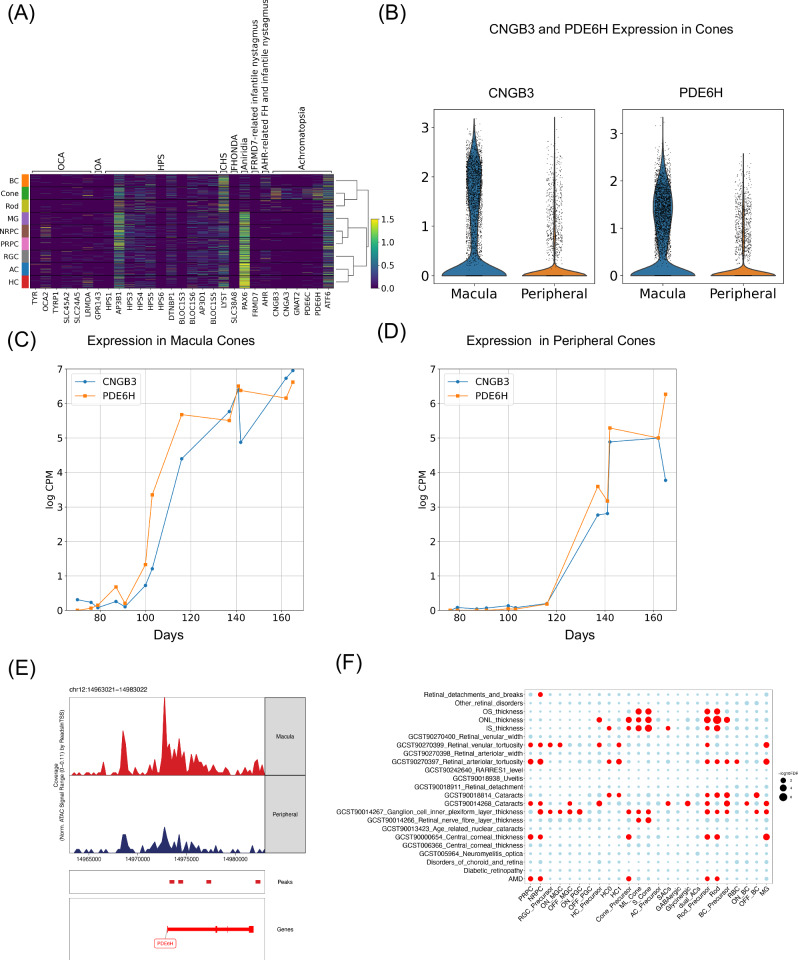
The development of the retina is under tight temporal and spatial control. To gain insights into the molecular basis of this process, we generate a single-nuclei dual-omic atlas of the human developing retina with approximately 220,000 nuclei from 14 human embryos and fetuses aged between 8 and 23-weeks post-conception with matched macular and peripheral tissues. This atlas captures all major cell classes in the retina, along with a large proportion of progenitors and cell-type-specific precursors. Cell trajectory analysis reveals a transition from continuous progression in early progenitors to a hierarchical development during the later stages of cell type specification. Both known and unrecorded candidate transcription factors, along with gene regulatory networks that drive the transitions of various cell fates, are identified. Comparisons between the macular and peripheral retinae indicate a largely consistent yet distinct developmental pattern. This atlas offers unparalleled resolution into the transcriptional and chromatin accessibility landscapes during development, providing an invaluable resource for deeper insights into retinal development and associated diseases.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Li, J. et al. Integrated multi-omics single cell atlas of the human retina. Preprint at bioRxiv10.21203/2Frs.3.rs-3471275/2Fv1 (2023).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
