

Massively parallel sample preparation for multiplexed single-cell proteomics using nPOP
- PMID: 39117766
- PMCID: PMC11614709
- DOI: 10.1038/s41596-024-01033-8
Massively parallel sample preparation for multiplexed single-cell proteomics using nPOP
Abstract
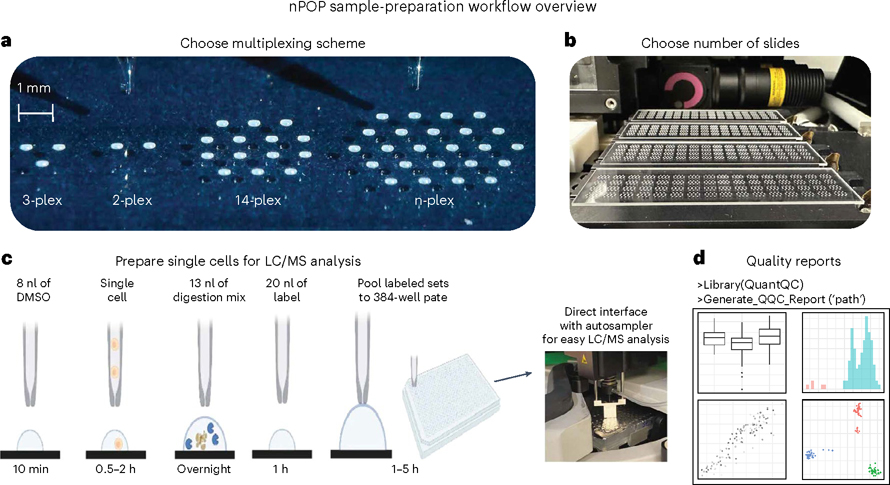
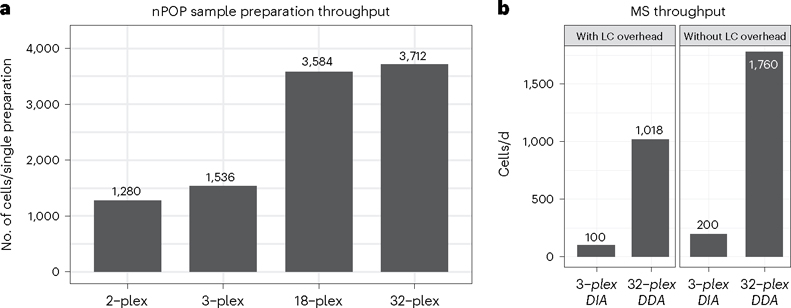
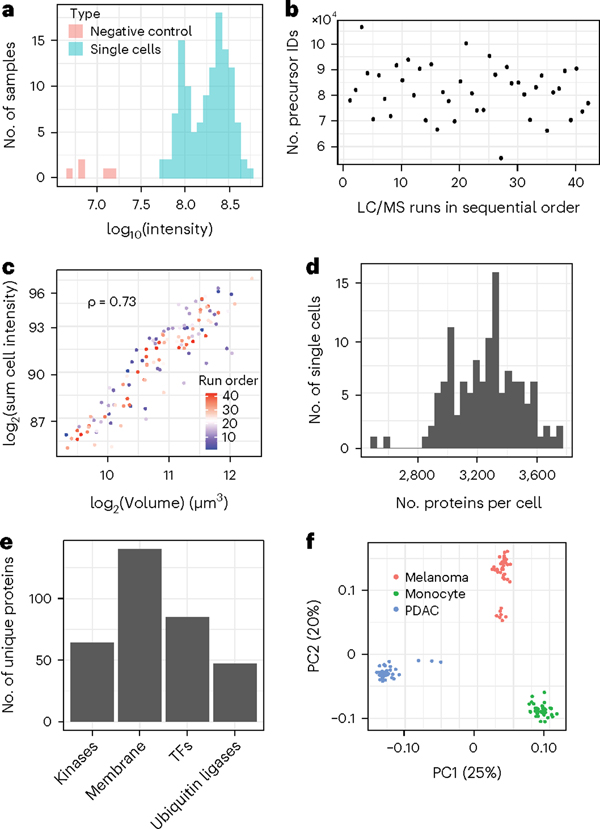
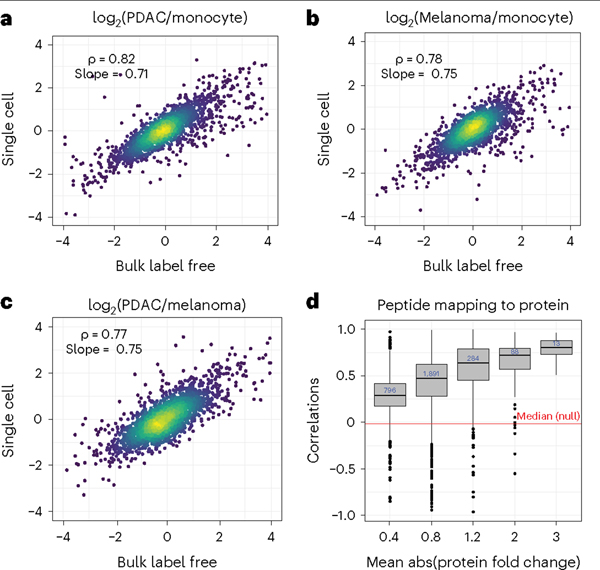
Single-cell proteomics by mass spectrometry (MS) allows the quantification of proteins with high specificity and sensitivity. To increase its throughput, we developed nano-proteomic sample preparation (nPOP), a method for parallel preparation of thousands of single cells in nanoliter-volume droplets deposited on glass slides. Here, we describe its protocol with emphasis on its flexibility to prepare samples for different multiplexed MS methods. An implementation using the plexDIA MS multiplexing method, which uses non-isobaric mass tags to barcode peptides from different samples for data-independent acquisition, demonstrates accurate quantification of ~3,000-3,700 proteins per human cell. A separate implementation with isobaric mass tags and prioritized data acquisition demonstrates analysis of 1,827 single cells at a rate of >1,000 single cells per day at a depth of 800-1,200 proteins per human cell. The protocol is implemented by using a cell-dispensing and liquid-handling robot-the CellenONE instrument-and uses readily available consumables, which should facilitate broad adoption. nPOP can be applied to all samples that can be processed to a single-cell suspension. It takes 1 or 2 d to prepare >3,000 single cells. We provide metrics and software (the QuantQC R package) for quality control and data exploration. QuantQC supports the robust scaling of nPOP to higher plex reagents for achieving reliable and scalable single-cell proteomics.
© 2024. Springer Nature Limited.
Conflict of interest statement
Competing interests: N.S. is a founding director and CEO of Parallel Squared Technology Institute, which is a nonprofit research institute. J.C. is an employee of SCIENION US Inc.
Figures


Update of
-
Massively parallel sample preparation for multiplexed single-cell proteomics using nPOP.bioRxiv [Preprint]. 2023 Nov 28:2023.11.27.568927. doi: 10.1101/2023.11.27.568927. bioRxiv. 2023. Update in: Nat Protoc. 2024 Dec;19(12):3750-3776. doi: 10.1038/s41596-024-01033-8. PMID: 38076795 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
