

CDK9 inhibition inhibits multiple oncogenic transcriptional and epigenetic pathways in prostate cancer
- PMID: 39117800
- PMCID: PMC11405875
- DOI: 10.1038/s41416-024-02810-8
CDK9 inhibition inhibits multiple oncogenic transcriptional and epigenetic pathways in prostate cancer
Erratum in
-
Author Correction: CDK9 inhibition constrains multiple oncogenic transcriptional and epigenetic pathways in prostate cancer.Br J Cancer. 2024 Nov;131(10):1719. doi: 10.1038/s41416-024-02862-w. Br J Cancer. 2024. PMID: 39448861 Free PMC article. No abstract available.
Abstract
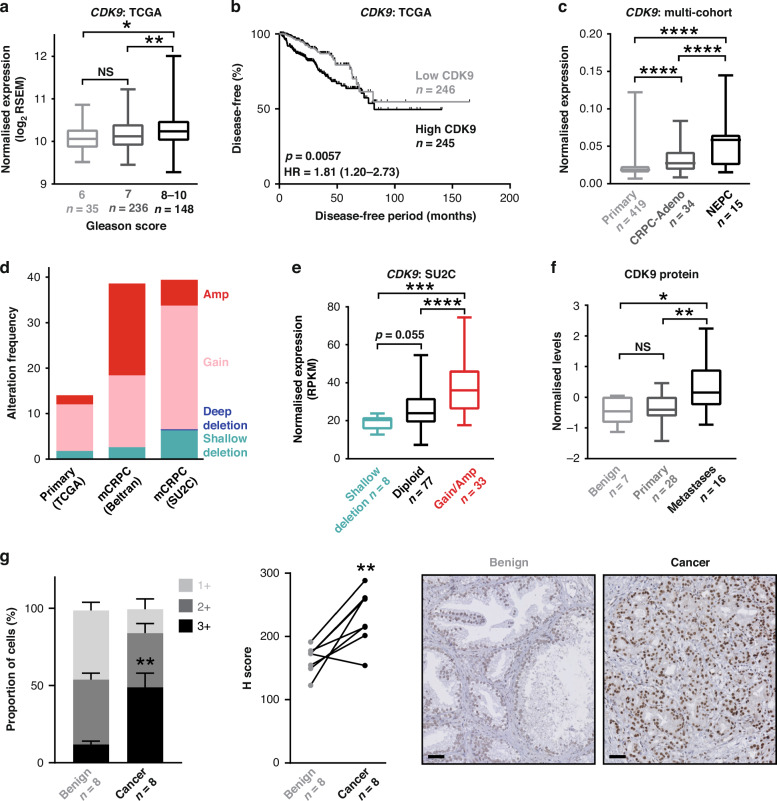
Background: Cyclin-dependent kinase 9 (CDK9) stimulates oncogenic transcriptional pathways in cancer and CDK9 inhibitors have emerged as promising therapeutic candidates.
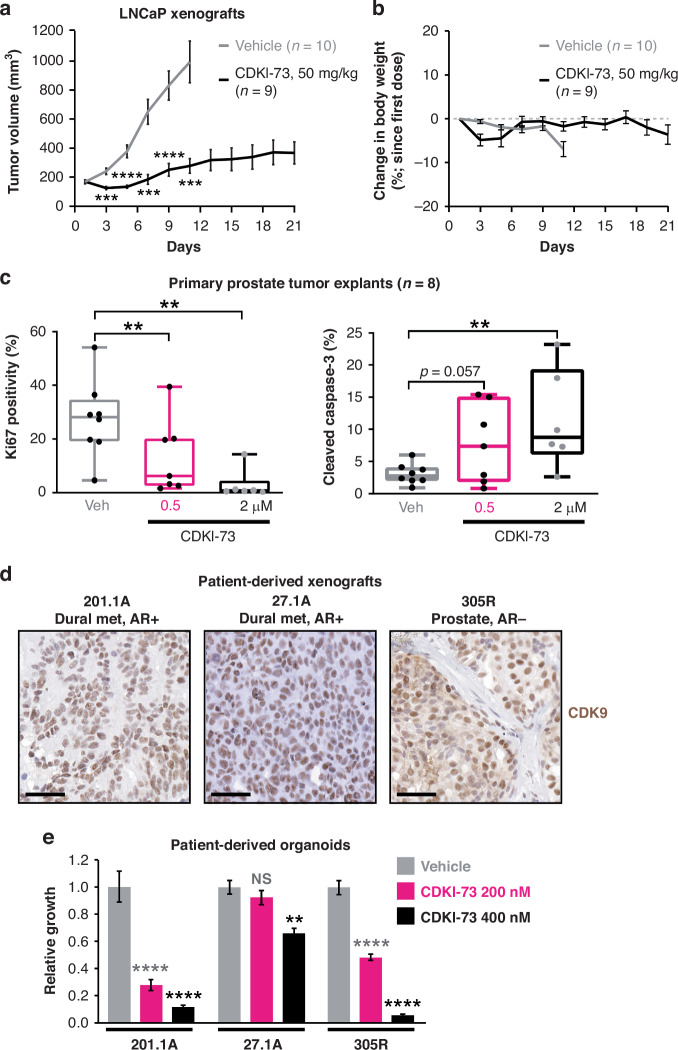
Methods: The activity of an orally bioavailable CDK9 inhibitor, CDKI-73, was evaluated in prostate cancer cell lines, a xenograft mouse model, and patient-derived tumor explants and organoids. Expression of CDK9 was evaluated in clinical specimens by mining public datasets and immunohistochemistry. Effects of CDKI-73 on prostate cancer cells were determined by cell-based assays, molecular profiling and transcriptomic/epigenomic approaches.
Results: CDKI-73 inhibited proliferation and enhanced cell death in diverse in vitro and in vivo models of androgen receptor (AR)-driven and AR-independent models. Mechanistically, CDKI-73-mediated inhibition of RNA polymerase II serine 2 phosphorylation resulted in reduced expression of BCL-2 anti-apoptotic factors and transcriptional defects. Transcriptomic and epigenomic approaches revealed that CDKI-73 suppressed signaling pathways regulated by AR, MYC, and BRD4, key drivers of dysregulated transcription in prostate cancer, and reprogrammed cancer-associated super-enhancers. These latter findings prompted the evaluation of CDKI-73 with the BRD4 inhibitor AZD5153, a combination that was synergistic in patient-derived organoids and in vivo.
Conclusion: Our work demonstrates that CDK9 inhibition disrupts multiple oncogenic pathways and positions CDKI-73 as a promising therapeutic agent for prostate cancer, particularly aggressive, therapy-resistant subtypes.
© 2024. The Author(s).
Conflict of interest statement
G Risbridger, R Taylor and M Lawrence (Research collaborations: AstraZeneca, Pfizer, Astellas, Zenith Epigenetics for research on compounds not associated with this study).
Figures




References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
