

Diesel exhaust particles alter mitochondrial bioenergetics and cAMP producing capacity in human bronchial epithelial cells
- PMID: 39118833
- PMCID: PMC11306203
- DOI: 10.3389/ftox.2024.1412864
Diesel exhaust particles alter mitochondrial bioenergetics and cAMP producing capacity in human bronchial epithelial cells
Abstract
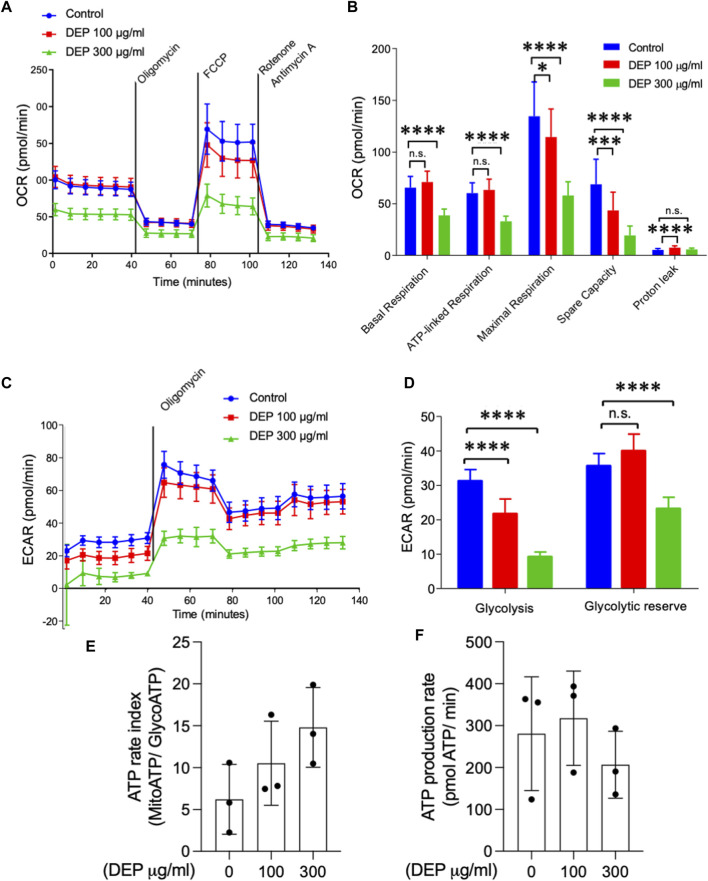
Introduction: Air pollution from diesel combustion is linked in part to the generation of diesel exhaust particles (DEP). DEP exposure induces various processes, including inflammation and oxidative stress, which ultimately contribute to a decline in lung function. Cyclic AMP (cAMP) signaling is critical for lung homeostasis. The impact of DEP on cAMP signaling is largely unknown. Methods: We exposed human bronchial epithelial (BEAS-2B) cells to DEP for 24-72 h and evaluated mitochondrial bioenergetics, markers of oxidative stress and inflammation and the components of cAMP signaling. Mitochondrial bioenergetics was measured at 72 h to capture the potential and accumulative effects of prolonged DEP exposure on mitochondrial function. Results: DEP profoundly altered mitochondrial morphology and network integrity, reduced both basal and ATP-linked respiration as well as the glycolytic capacity of mitochondria. DEP exposure increased gene expression of oxidative stress and inflammation markers such as interleukin-8 and interleukin-6. DEP significantly affected mRNA levels of exchange protein directly activated by cAMP-1 and -2 (Epac1, Epac2), appeared to increase Epac1 protein, but left phospho-PKA levels unhanged. DEP exposure increased A-kinase anchoring protein 1, β2-adrenoceptor and prostanoid E receptor subtype 4 mRNA levels. Interestingly, DEP decreased mRNA levels of adenylyl cyclase 9 and reduced cAMP levels stimulated by forskolin (AC activator), fenoterol (β2-AR agonist) or PGE2 (EPR agonist). Discussion: Our findings suggest that DEP induces mitochondrial dysfunction, a process accompanied by oxidative stress and inflammation, and broadly dampens cAMP signaling. These epithelial responses may contribute to lung dysfunction induced by air pollution exposure.
Keywords: air pollution; cAMP; diesel exhaust particles; lung; mitochondria; oxidative stress.
Copyright © 2024 Cattani-Cavalieri, Trombetta-Lima, Yan, Manzano-Covarrubias, Baarsma, Oun, van der Veen, Oosterhout, Dolga, Ostrom, Valenca and Schmidt.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures






Similar articles
-
Multi-cellular human bronchial models exposed to diesel exhaust particles: assessment of inflammation, oxidative stress and macrophage polarization.Part Fibre Toxicol. 2018 May 2;15(1):19. doi: 10.1186/s12989-018-0256-2. Part Fibre Toxicol. 2018. PMID: 29716632 Free PMC article.
-
Diesel exhaust particles induced release of interleukin 6 and 8 by (primed) human bronchial epithelial cells (BEAS 2B) in vitro.Exp Lung Res. 1998 Jan-Feb;24(1):85-100. doi: 10.3109/01902149809046056. Exp Lung Res. 1998. PMID: 9457471
-
α-Bisabolol alleviates diesel exhaust particle-induced lung injury and mitochondrial dysfunction by regulating inflammatory, oxidative stress, and apoptotic biomarkers through the c-Jun N-terminal kinase signaling pathway.Front Pharmacol. 2025 Jan 3;15:1485101. doi: 10.3389/fphar.2024.1485101. eCollection 2024. Front Pharmacol. 2025. PMID: 39830335 Free PMC article.
-
Nanodomains in cardiopulmonary disorders and the impact of air pollution.Biochem Soc Trans. 2020 Jun 30;48(3):799-811. doi: 10.1042/BST20190250. Biochem Soc Trans. 2020. PMID: 32597478 Free PMC article. Review.
-
Diesel exhaust particles activate human bronchial epithelial cells to express inflammatory mediators in the airways: a review.Respirology. 2000 Jun;5(2):197-203. doi: 10.1046/j.1440-1843.2000.00245.x. Respirology. 2000. PMID: 10894110 Review.
Cited by
-
Regulation of Airway Epithelial-Derived Alarmins in Asthma: Perspectives for Therapeutic Targets.Biomedicines. 2024 Oct 11;12(10):2312. doi: 10.3390/biomedicines12102312. Biomedicines. 2024. PMID: 39457624 Free PMC article. Review.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials