

Exploring 2-Sulfonylpyrimidine Warheads as Acrylamide Surrogates for Targeted Covalent Inhibition: A BTK Story
- PMID: 39119945
- PMCID: PMC11345841
- DOI: 10.1021/acs.jmedchem.3c01927
Exploring 2-Sulfonylpyrimidine Warheads as Acrylamide Surrogates for Targeted Covalent Inhibition: A BTK Story
Abstract
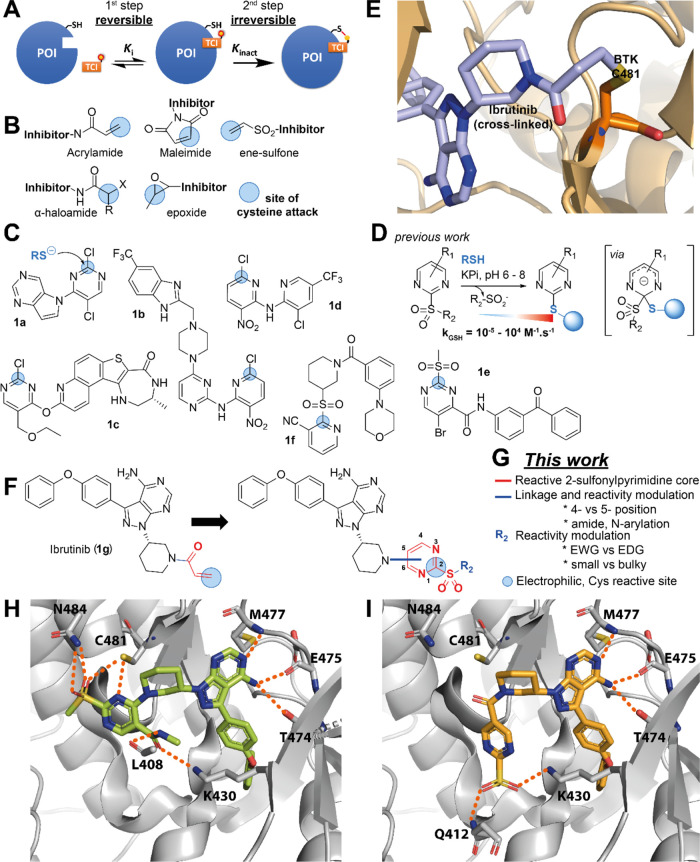
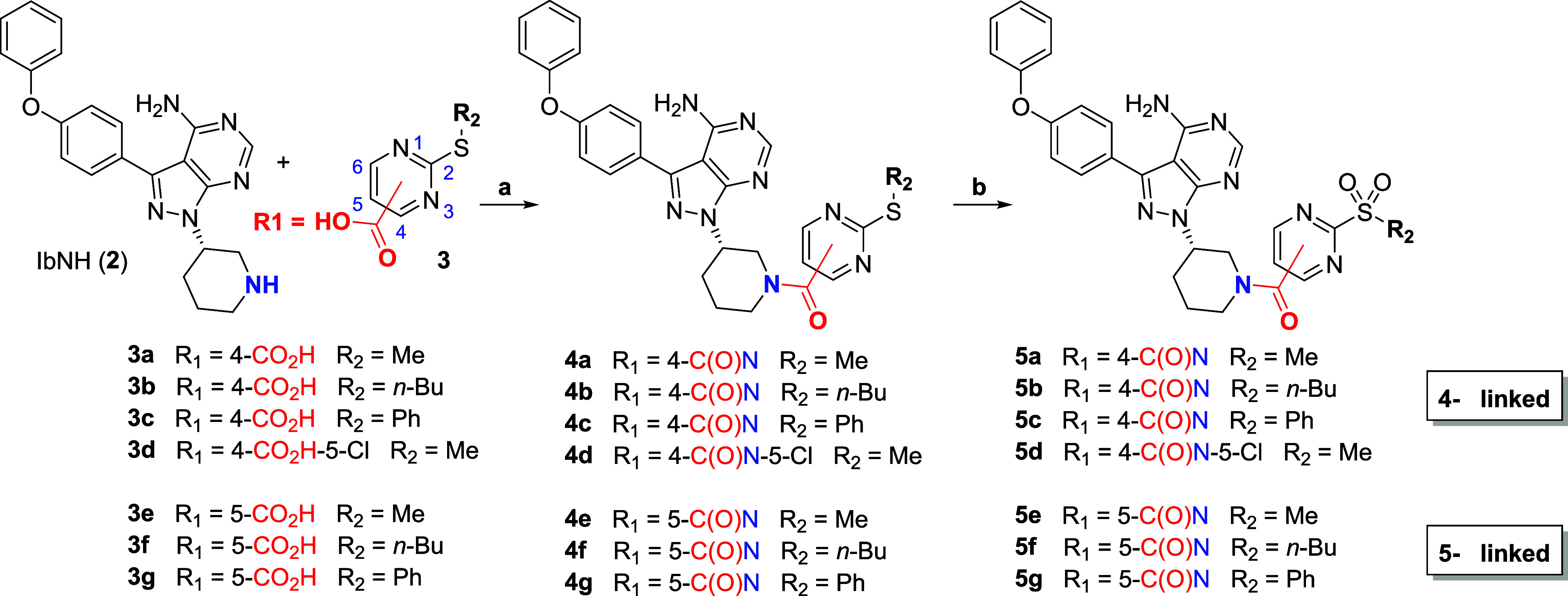
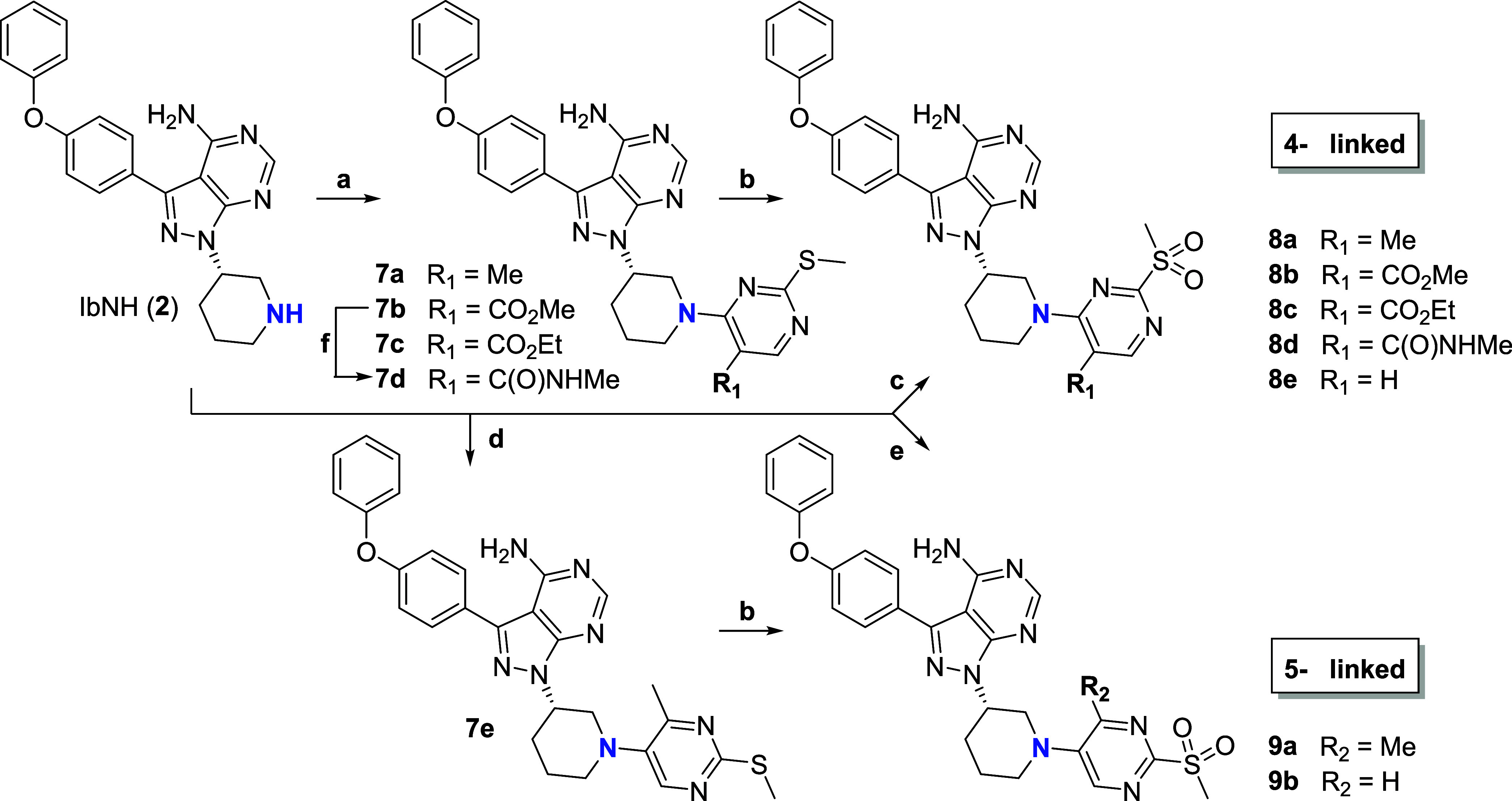
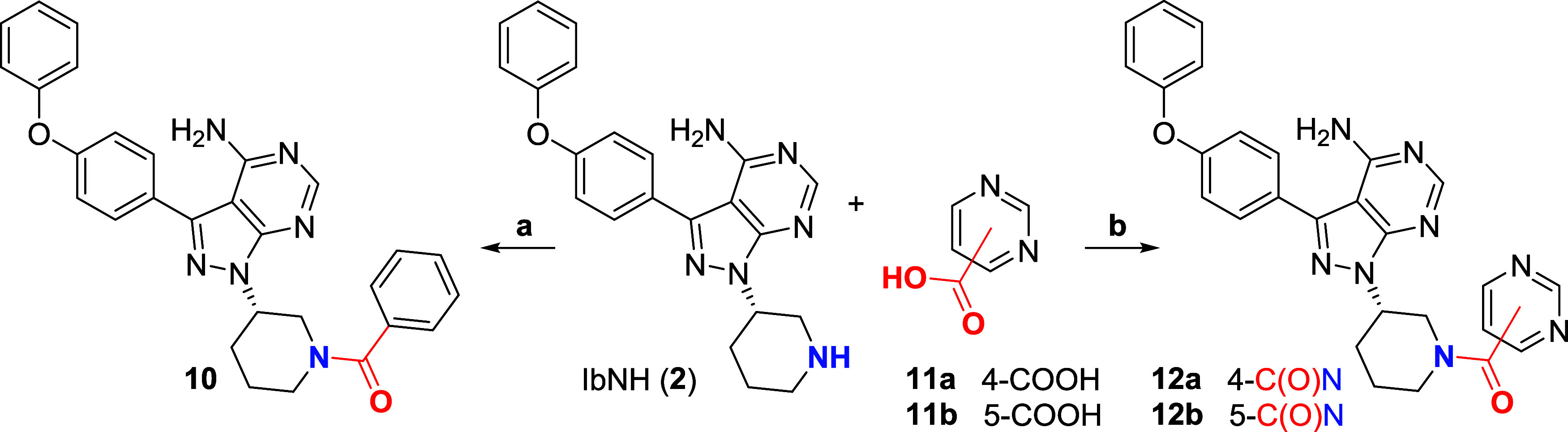
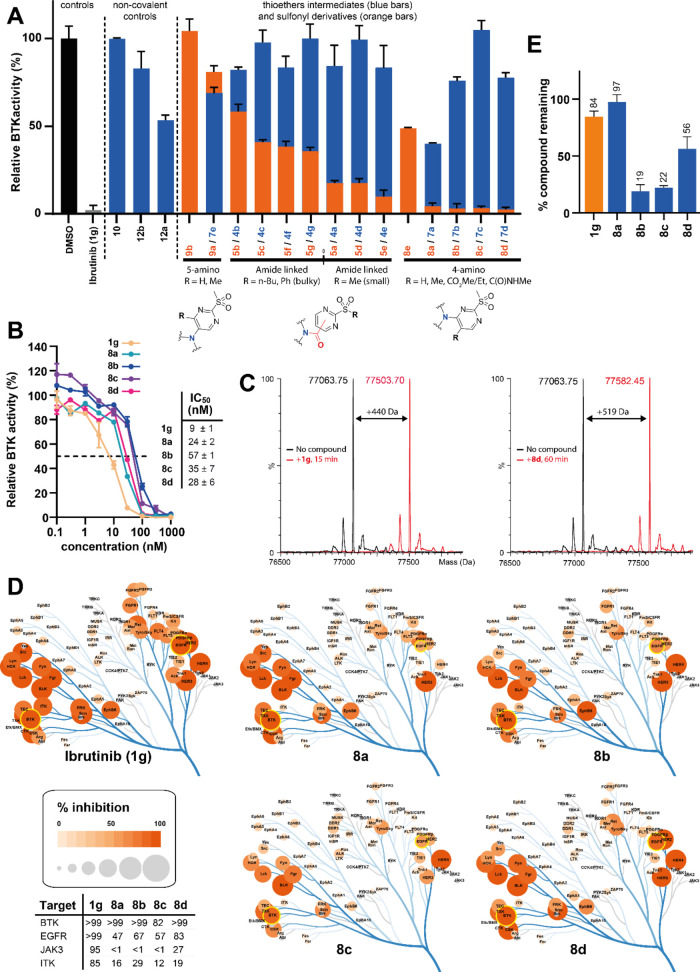
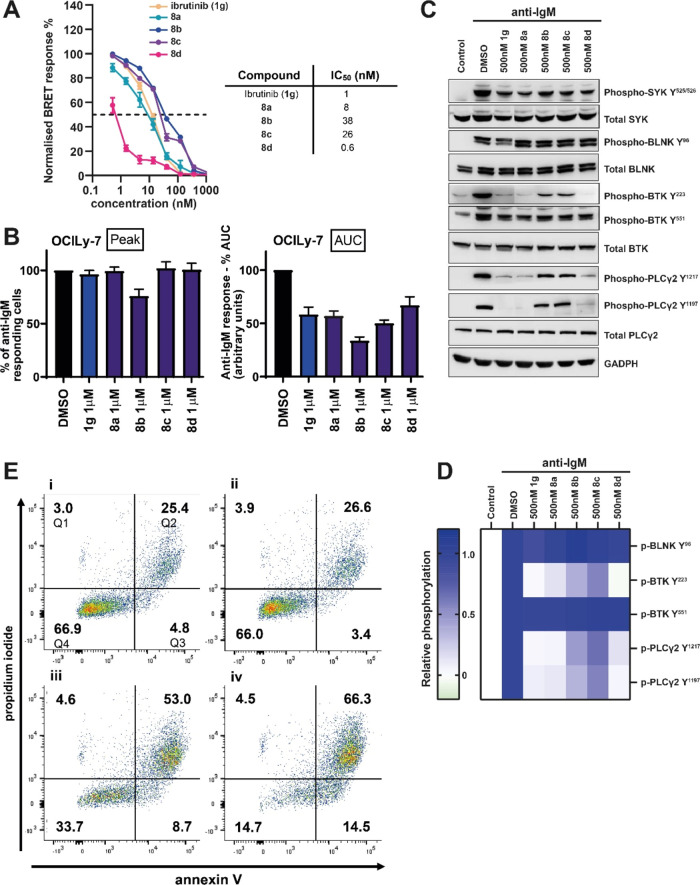
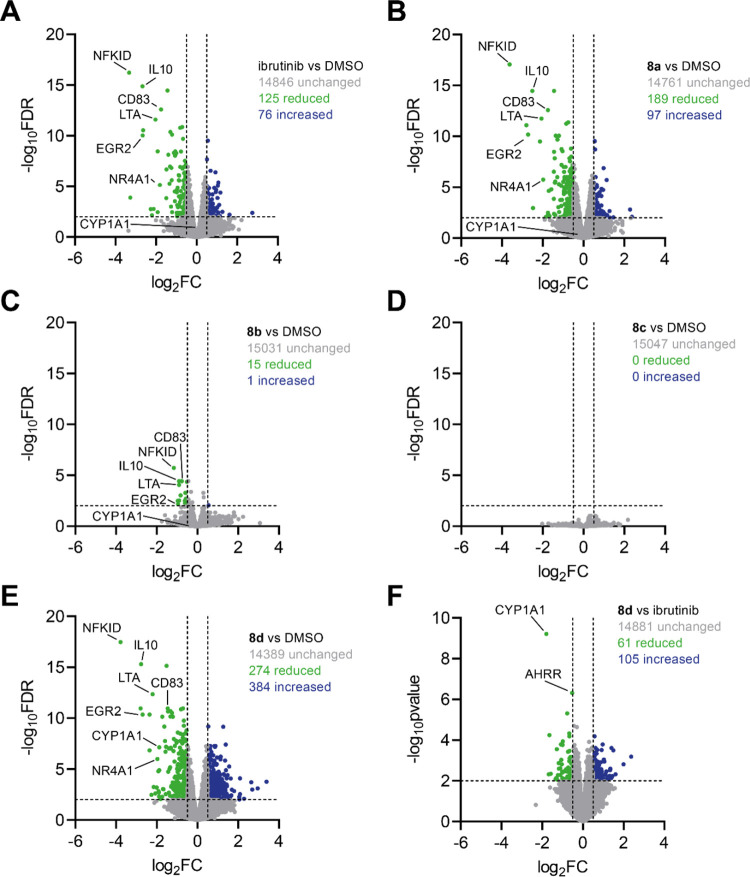
Targeted covalent inhibitors (TCIs) directing cysteine have historically relied on a narrow set of electrophilic "warheads". While Michael acceptors remain at the forefront of TCI design strategies, they show variable stability and selectivity under physiological conditions. Here, we show that the 2-sulfonylpyrimidine motif is an effective replacement for the acrylamide warhead of Ibrutinib, for the inhibition of Bruton's tyrosine kinase. In a few iterations, we discovered new derivatives, which inhibit BTK both in vitro and in cellulo at low nanomolar concentrations, on par with Ibrutinib. Several derivatives also displayed good plasma stability and reduced off-target binding in vitro across 135 tyrosine kinases. This proof-of-concept study on a well-studied kinase/TCI system highlights the 2-sulfonylpyrimidine group as a useful acrylamide replacement. In the future, it will be interesting to investigate its wider potential for developing TCIs with improved pharmacologies and selectivity profiles across structurally related protein families.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
