

This is a preprint.
Insertion and Anchoring of HIV-1 Fusion Peptide into Complex Membrane Mimicking Human T-cell
- PMID: 39131401
- PMCID: PMC11312619
- DOI: 10.1101/2024.08.02.606381
Insertion and Anchoring of HIV-1 Fusion Peptide into Complex Membrane Mimicking Human T-cell
Update in
-
Insertion and Anchoring of the HIV-1 Fusion Peptide into a Complex Membrane Mimicking the Human T-Cell.J Phys Chem B. 2024 Dec 26;128(51):12710-12727. doi: 10.1021/acs.jpcb.4c05018. Epub 2024 Dec 13. J Phys Chem B. 2024. PMID: 39670799
Abstract
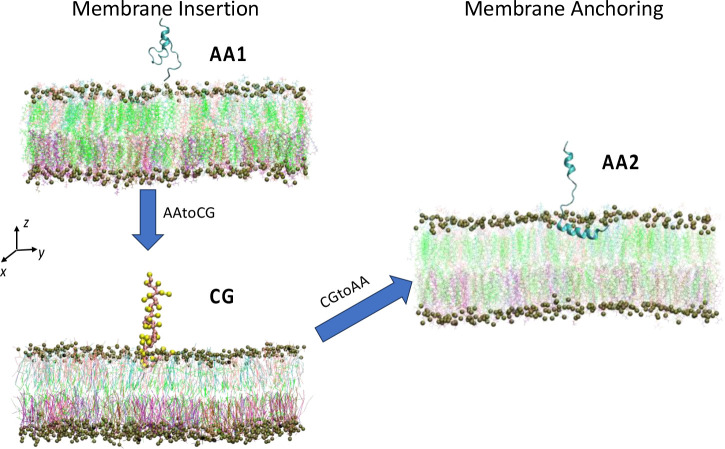
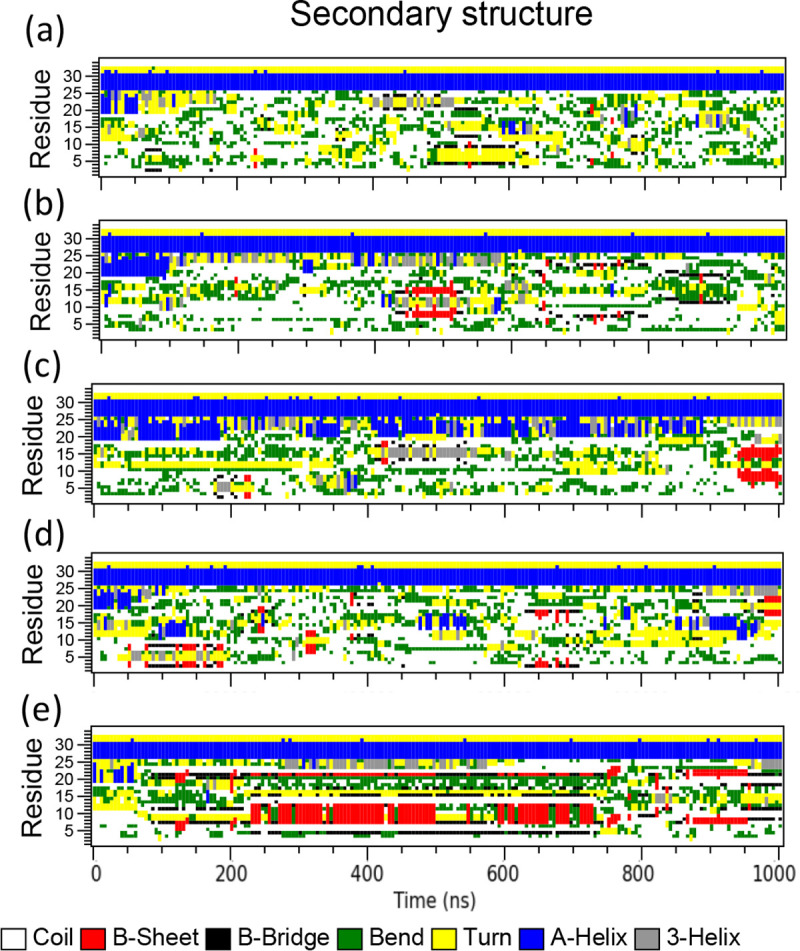
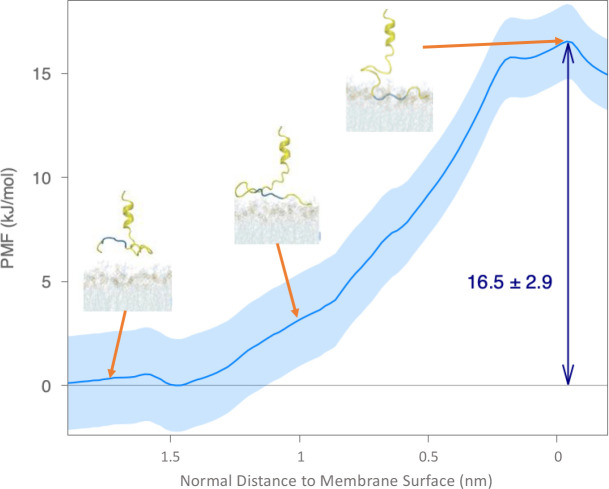
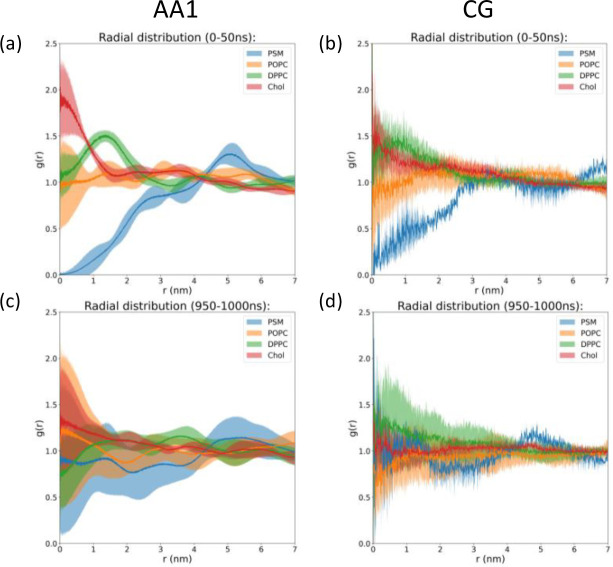
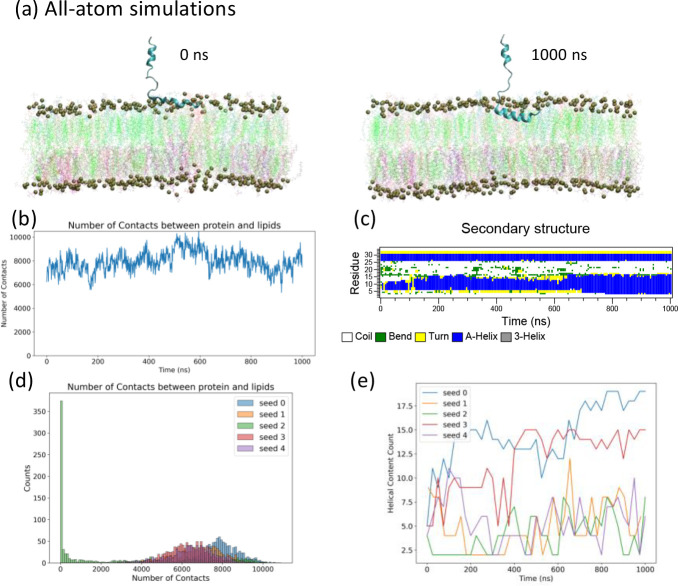
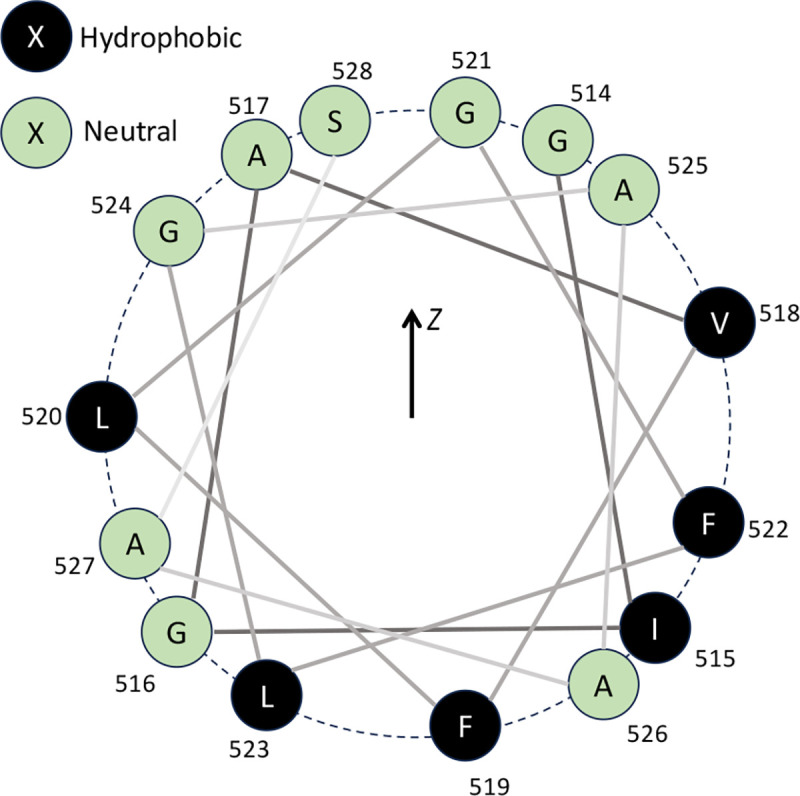
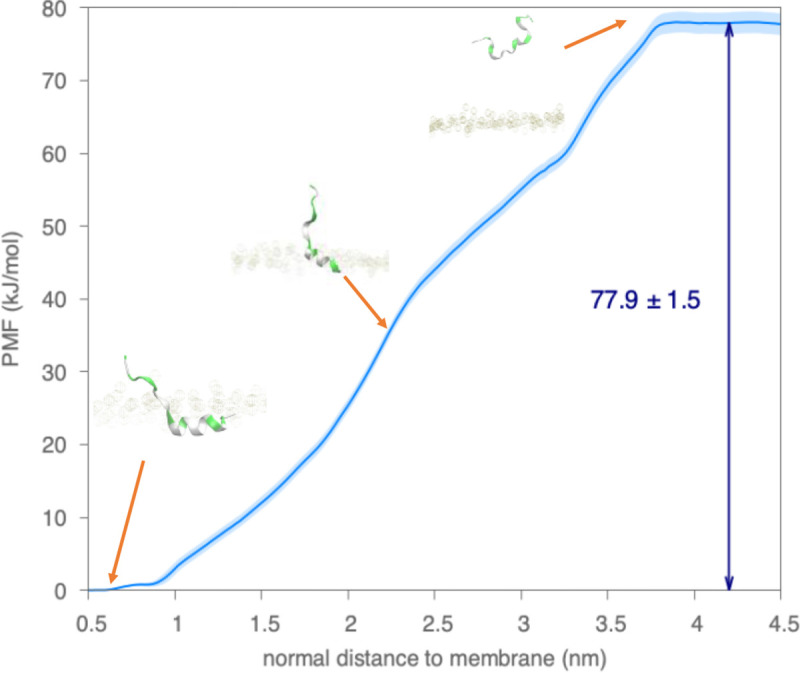
A fundamental understanding of how HIV-1 envelope (Env) protein facilitates fusion is still lacking. The HIV-1 fusion peptide, consisting of 15 to 22 residues, is the N-terminus of the gp41 subunit of the Env protein. Further, this peptide, a promising vaccine candidate, initiates viral entry into target cells by inserting and anchoring into human immune cells. The influence of membrane lipid reorganization and the conformational changes of the fusion peptide during the membrane insertion and anchoring processes, which can significantly affect HIV-1 cell entry, remains largely unexplored due to the limitations of experimental measurements. In this work, we investigate the insertion of the fusion peptide into an immune cell membrane mimic through multiscale molecular dynamics simulations. We mimic the native T-cell by constructing a 9-lipid asymmetric membrane, along with geometrical restraints accounting for insertion in the context of gp41. To account for the slow timescale of lipid mixing while enabling conformational changes, we implement a protocol to go back and forth between atomistic and coarse-grained simulations. Our study provides a molecular understanding of the interactions between the HIV-1 fusion peptide and the T-cell membrane, highlighting the importance of conformational flexibility of fusion peptides and local lipid reorganization in stabilizing the anchoring of gp41 into the targeted host membrane during the early events of HIV-1 cell entry. Importantly, we identify a motif within the fusion peptide critical for fusion that can be further manipulated in future immunological studies.
Keywords: HIV-1; coarse-grained simulations; complex membrane; fusion peptide; molecular dynamics simulation; multiscale modeling; viral cell entry.
Figures






References
-
- Gorai B., et al. , Concerted Interactions between Multiple gp41 Trimers and the Target Cell Lipidome May Be Required for HIV-1 Entry. J Chem Inf Model, 2021. 61(1): p. 444–454. - PubMed
-
- Heller W.T., A small-angle neutron scattering study of the physical mechanism that drives the action of a viral fusion peptide. Chem Phys Lipids, 2021. 234: p. 105022. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources